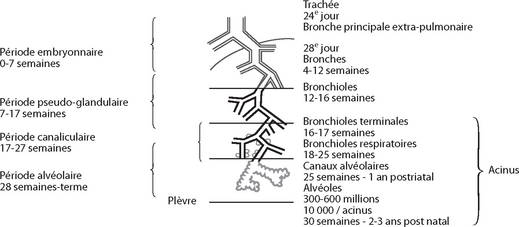
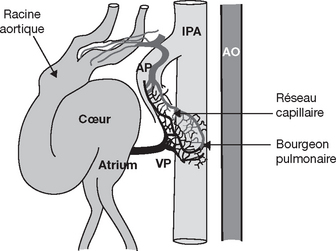
CHAPITRE 4 MALFORMATIONS CONGÉNITALES PULMONAIRES 4.1 MALFORMATIONS CONGÉNITALES RESPIRATOIRES DE L’ADULTE P. Lacombe, M. El Hajjam, J. Sellier, A. Machet, S. Binsse, C. Lagrange, S. Chagnon, J.-P. Pelage and C. Beigelman Il n’est pas rare de rencontrer des malformations congénitales respiratoires à l’âge adulte. Or, dans cette tranche d’âge, les praticiens en charge du patient ont tendance à ne pas les suspecter, ce qui, malgré les progrès de l’imagerie non invasive, peut déboucher sur la réalisation d’actes invasifs (endoscopies, angiographies) parfois inutiles. Ces malformations peuvent être découvertes de façon fortuite à l’occasion d’un examen pratiqué pour une autre raison ou sous le masque d’une complication chronique, parfois trompeuse, ou aiguë, qu’il conviendra de rapporter à sa véritable étiologie [61, 82]. L’extraordinaire diversité des cas publiés illustre la grande hétérogénéité de ces malformations, souvent associées à des anomalies cardiovasculaires, squelettiques, abdominales ou diaphragmatiques. Les diverses tentatives de classifications unicistes, fondées sur de grandes séries cliniques et sur l’embryologie, se sont heurtées à un nombre illimité de variantes et n’ont fait que renforcer la confusion [10]. Il n’en reste pas moins vrai que le support embryologique est indispensable pour tenter d’élucider la pathogénie de ces malformations [29]. Nous nous intéresserons successivement aux anomalies du développement pulmonaire ou lobaire, regroupées sous le nom de « poumon dysmorphique », puis aux anomalies trachéo-broncho-pulmonaires et œsophagiennes, depuis les variantes de la normale jusqu’aux malformations les plus complexes. Nous ne ferons qu’énumérer les principales malformations qui se manifestent presque exclusivement en période néonatale ou pédiatrique précoce [7]. Les malformations congénitales des vaisseaux pulmonaires et les séquestrations sont traitées dans le sous-chapitre suivant (4.2). Le développement pulmonaire s’effectue en deux étapes principales, intra-utérine et postnatale. Le développement intra-utérin peut se scinder en quatre périodes distinctes [30, 84] (fig. 4-1). La première période, ou « période embryonnaire », débute au 26e jour pour se terminer à la 7e semaine. Dès le début de cette période apparaît, dans la partie ventrale de l’intestin primitif, un diverticule se situant à la jonction entre ses segments occipitaux et cervicaux. Il est limité par un épithélium endodermique et s’étend au sein du mésenchyme splanchnique environnant. Sa croissance se fait en direction caudale, aboutissant au bout de 2 jours à la formation des bourgeons pulmonaires primitifs droit, à direction caudale, et gauche, à direction plus transversale. Le bourgeon droit apparaît juste avant le bourgeon gauche. Ce diverticule ventral respiratoire se sépare progressivement de sa composante œsophagienne dorsale, en raison de la croissance vers la ligne médiane de deux crêtes du mésoderme qui s’unissent pour former une véritable cloison entre l’œsophage primitif et les bourgeons bronchopulmonaires. Il s’agit du septum trachéo-œsophagien. Fig. 4-1 Illustration du développement pré- et postnatal de chacune des structures aériennes pulmonaires en fonction du temps. Source : modifie d’après Hislop et al. [30]. Les bourgeons pulmonaires, situés de part et d’autre de l’œsophage primitif, s’allongent progressivement au sein du mésenchyme pour constituer les sacs pulmonaires primitifs qui se subdivisent initialement en cinq bourgeons lobaires, trois à droite et deux à gauche, puis en dix bourgeons segmentaires, entièrement constitués à la 7e semaine. À la fin de cette période, les deux poumons sont ainsi séparés et parfaitement distincts de la masse cardiopéricardique située en avant [53]. La dernière période intra-utérine, dite « alvéolaire », va de la 27e semaine au terme de la grossesse. Les acini sont bien développés à ce stade. Les alvéoles apparaissent dès la 29e semaine dans la paroi des saccules, sous forme d’indentations ou de septa. À la 34e semaine, les alvéoles ont un aspect cupuliforme avec une double couche de capillaires sous les cellules épithéliales. À terme, entre le tiers et la moitié du nombre des alvéoles présents à l’âge adulte sont formés. Chez les prématurés, les anomalies du développement sont à l’origine de dysplasies bronchopulmonaires, sources de troubles respiratoires parfois graves. L’évolution à long terme de ces dysplasies est maintenant mieux connue [20, 24]. Nous distinguerons artificiellement le développement des vaisseaux périphériques et des vaisseaux centraux. En effet, l’évolution embryonnaire des portions proximales et distales des vaisseaux pulmonaires se fait de façon différente mais synchrone (fig. 4-2). Le développement des vaisseaux bronchiques sera évoqué ensuite [30]. Fig. 4-2 Illustration schématique du développement des vaisseaux pulmonaires résultant d’une reconstruction de coupes sériées d’un embryon humain de 34 jours. Source : modifie d’après Hislop et al. [30]. Reconstruction Susan Hall. À partir d’études expérimentales récentes, il a été possible, grâce à un immunomarquage de cellules endothéliales et musculaires lisses, de découvrir dès le 28e jour des groupes de cellules endothéliales autour des bourgeons bronchiques primitifs au sein du mésenchyme [13]. Au 34e jour, ces cellules s’organisent en plexus capillaires (plexus de Huntington) alimentés primitivement par des artères segmentaires ventrales de l’aorte dorsale et se drainant vers les veines systémiques cardinales et ombilicovitellines. Autour de chaque nouveau bourgeon bronchique et à une distance déterminée de l’épithélium, apparaissent des structures tubulaires dont la coalescence s’effectue longitudinalement par rapport aux voies aériennes. Ces structures tubulaires sont à l’origine de la constitution des artères et des veines pulmonaires périphériques primitives. La fusion des plexus et de ces structures tubulaires constitue l’ensemble du réseau vasculaire pulmonaire primitif périphérique. Initialement, artères et veines sont situées à égale distance des voies aériennes. Progressivement, si les artères restent proches des structures aériennes, les veines s’en séparent pour rejoindre leur topographie définitive périlobulaire. Une telle distribution explique peut-être la différence de structure entre les parois artérielles et veineuses : les parois artérielles sont investies par des cellules musculaires lisses dérivées des bourgeons bronchiques adjacents en cours de développement pour constituer leur tunique profonde. À l’inverse, les parois veineuses sont constituées principalement de cellules fibroblastiques provenant du mésenchyme pulmonaire environnant [30]. Il existe deux hypothèses concernant l’évolution embryologique de la vascularisation bronchique : – disparition de la vascularisation systémique des plexus de Huntington provenant de l’aorte à l’exception des artères bronchiques ; – disparition totale de la circulation provenant de l’aorte dorsale sus-cœliaque ; cette disparition totale serait suivie de l’apparition secondaire vers la 9e-10e semaine de bourgeons vasculaires provenant de la face antérieure de l’aorte descendante ou des artères intercostales avec croissance vers l’arbre trachéobronchique puis connexion secondaire aux vaisseaux pulmonaires [30] ; les séquestrations pulmonaires, dans ce cas, seraient liées à la persistance anormale des artères systémiques primitives entraînées parfois dans la région sous-diaphragmatique en raison du glissement caudal du tronc cœliaque lors du développement en longueur de l’embryon. Sur le plan physiopathologique un défaut de développement des structures artérielles pulmonaires, comme dans l’atrésie pulmonaire, induit un développement compensateur des vaisseaux bronchiques. – celles qui reposent sur l’embryologie et l’anatomie [10] ; selon la date de survenue de la lésion causale sur le bourgeon bronchopulmonaire, on constatera des anomalies du développement ou de la croissance des structures respiratoires d’aspect et de topographie différents ; si ce type de classification apparaît possible pour les malformations simples comme les dérivés kystiques de l’intestin primitif antérieur et les atrésies bronchiques, à l’inverse, les explications embryologiques s’avèrent beaucoup plus complexes pour des malformations multitissulaires comme les séquestrations extralobaires et les retours veineux pulmonaires anormaux ; – des classifications à partir de la morphologie ont été proposées [26] ; dans ce cas, les malformations sont interprétées comme un continuum entre une anomalie vasculaire et une anomalie pulmonaire pures, avec un groupe de malformations intermédiaires où les deux composantes sont associées à des degrés variables ; – le troisième type de classification fait référence à des cadres radiomorphologiques [54] ; il est subdivisé en deux groupes : en cas de trouble de développement d’un poumon ou d’un lobe, on parlera de « poumon dysmorphique » ; dans les autres cas, on parlera de « malformation focale trachéo-broncho-pulmonaire et œsophagienne ». Nous adopterons cette dernière classification radiomorpho-logique car la radiographie thoracique standard permet une distinction facile entre les deux groupes pathologiques. De plus, les indications thérapeutiques sont en apparence simplifiées : une malformation focale fait opter plus volontiers pour un traitement chirurgical [54]. Ce groupe d’anomalies congénitales recouvre la majorité des défauts de développement embryonnaire des structures pulmonaires et vasculaires pouvant affecter l’ensemble d’un poumon ou se limiter à un ou plusieurs lobes (fig. 4-3). Initialement décrit en se référant spécifiquement aux données de l’angiographie, le poumon dysmorphique est beaucoup mieux connu depuis que le scanner et, à un moindre degré, l’IRM ont permis une meilleure analyse de la composante parenchymateuse [53, 57]. Nous distinguerons deux groupes principaux selon que le défaut de développement affecte tout un poumon ou une partie seulement de ce dernier. Fig. 4-3 Malformation bronchopulmonaire et vasculaire découverte à l’âge adulte. De même, certaines agénésies ont un pronostic grave, à expression néonatale exclusive, quand elles s’intègrent dans des complexes malformatifs associant des anomalies vertébrales (bloc vertébral, hémivertèbre), une imperforation anale, des malformations cardiaques, des fistules œsotrachéales, des lésions rénales et des extrémités, regroupés sous le vocable « syndrome VACTERL » ou «association VATER » [37, 72]. Certaines observations illustrent des malformations des arcs branchiaux et des membres, homolatérales à l’atteinte pulmonaire [12]. Les agénésies bilatérales sont incompatibles avec la vie. L’image d’allure pleurale chronique, initialement considérée comme un tissu de comblement venant compenser l’agénésie pulmonaire est, d’après la tomodensitométrie, le résultat de l’accumulation du tissu graisseux sous-pleural là où le déplacement du médiastin et la hernie du parenchyme controlatéral ne peuvent pas occuper l’espace laissé vacant. La clarté rétrosternale de profil est l’expression du passage transmédiastinal antérieur du poumon controlatéral en expansion compensatrice [66]. Elle se caractérise par la réduction du nombre ou de la taille des espaces aériens, des vaisseaux et des alvéoles. Elle est visible en imagerie par un poumon de taille réduite mais morphologiquement harmonieux. Cette hypoplasie primaire doit être distinguée de l’hypoplasie secondaire à l’existence d’une masse à développement intrathoracique (hernie diaphragmatique, tumeur, épanchement pleural), à des anomalies morphologiques de la cage thoracique ou aux conséquences d’un oligohydramnios [36]. Une forme particulière a été décrite dans laquelle le poumon hypoplasique se caractérise par l’absence de structures bronchiolo-alvéolaires : l’hypoplasie globale kystique (fig. 4-4). Fig. 4-4 Hypoplasie kystique pulmonaire droite. Coupe de 1 mm en fenêtre parenchymateuse pulmonaire. Elle se définit par l’absence de toute structure bronchopulmonaire et vasculaire d’un lobe ou plusieurs lobes (fig. 4-5 et 4-6). Fig. 4-5 Agénésie lobaire supérieure et moyenne droite, hypoplasie lobaire inférieure. Fig. 4-6 Agénésie du culmen. Tomodensitométrie axiale en coupes fines avec injection d’iode. Dans l’aplasie lobaire, il existe un cul-de-sac bronchique souvent ectasique entouré d’éléments parenchymateux peu différenciés pouvant communiquer avec des cavités kystiques (fig. 4-7). Les cavités kystiques ont un aspect très proche de celui des bronchectasies et correspondent à un trouble de développement bronchiolo-alvéolaire. Les vaisseaux pulmonaires sont soit très grêles, soit absents. Une composante systémique est possible. Fig. 4-7 Aplasie lobaire supérieure droite B1-B3 dans un cas de lévo-isomérisme. Ici le pédicule bronchique lobaire existe et présente des ramifications multiples. Celles-ci peuvent communiquer avec des cavités kystiques correspondant au défaut de développement bronchiolo-alvéolaire. Les artères pulmonaires sont généralement de petite taille et associées à des artères systémiques (fig. 4-8). Fig. 4-8 Hypoplasie harmonieuse pulmonaire droite. L’agénésie, l’aplasie ou l’hypoplasie d’un ou plusieurs lobes est dénommée « syndrome du poumon hypogénétique » [53, 78]. En radiographie standard, le syndrome du poumon hypo-génétique se manifeste presque exclusivement à droite. Les cas intéressant le côté gauche sont exceptionnels [63]. L’hémithorax droit apparaît réduit de volume avec un déplacement cardiomédiastinal vers la droite et un aspect flou du contour droit de la silhouette cardiaque. L’aspect du hile droit, lorsque celui-ci n’est pas masqué par la rotation cardiomédiastinale, ressemble parfois au hile gauche avec une bronche hyparté-rielle, signifiant alors l’association à un lévo-isomérisme. L’incidence de profil retrouve une bande opaque rétrosternale liée à une diminution du diamètre antéro-postérieur du thorax du côté du poumon hypogénétique. L’angioscanner hélicoïdal permet d’optimiser l’analyse des anomalies bronchopulmonaires, vasculaires et diaphragmatiques [53]. Les lobes le plus souvent atteints par l’hypoplasie sont le lobe supérieur droit et le lobe moyen. Les modifications scissurales sont très variables. L’association à un diaphragme accessoire ou à une hernie diaphragmatique est classique. Le scanner hélicoïdal est réalisé en collimation fine avec un intervalle de reconstruction proche de la moitié de l’épaisseur de coupe, de façon idéale en 1,25 mm/0,6 mm. Il permet de faire la distinction entre une agénésie lobaire dépourvue de tout élément bronchique et donc sans risque de complication infectieuse, et une aplasie ou une hypoplasie compliquée d’« ectasies bronchiques », sources possibles de suppurations et de saignements. En contrepartie, le scanner fait difficilement la distinction entre des images de dilatation des bronches acquises et celles d’une aplasie bronchiolo-alvéolaire ou d’une hypoplasie lobaire congénitale dysharmo-nieuse. Le point essentiel est de faire un bilan topographique à visée préopératoire. L’angioscanner permet d’optimiser l’analyse des voies artérielles et veineuses pulmonaires, et de détecter et faire la cartographie des fréquentes participations systémiques dans le poumon droit hypogénétique (fig. 4-9). La présence d’un retour veineux pulmonaire anormal permet d’intégrer le syndrome du poumon hypogénétique dans celui du syndrome veinolobaire pulmonaire congénital. Fig. 4-9 Hypoplasie droite et malformations vasculaire et diaphragmatique. Ce syndrome malformatif associe les éléments du poumon droit hypogénétique à un retour veineux pulmonaire anormal, partiel ou total, vers la veine cave inférieure sous-diaphragmatique, une veine hépatique, l’atrium droit, le sinus coronaire, la veine azygos ou, plus rarement, la veine cave supérieure et la veine porte. Le siège à gauche est très rare [50]. La pathogénie de ce syndrome est mal connue [57]. Woodring et al. [78] en ont rapporté 29 cas. L’hypoplasie touchait préférentiellement le lobe moyen (65 %), puis le lobe supérieur droit (40 %), un lobe inférieur (20 %) et plusieurs lobes dans 45 % des cas. Dans 38 % des cas, il s’agissait d’un lobe hypoplasique et, dans 62 % des cas, d’une aplasie ou d’une agénésie. On oppose les formes asymptomatiques à celles révélées par une dyspnée d’effort en rapport avec l’hypoplasie, par une suppuration bronchique similaire à celle du poumon droit hypogénétique et par le shunt gauche-droite lié au retour veineux pulmonaire anormal. Ce dernier ne devient symptomatique que dans la mesure où le rapport entre le débit pulmonaire et le débit systémique est supérieur ou égal à 2 ou si le shunt se complique d’hypertension artérielle pulmonaire (fig. 4-10) [28]. Fig. 4-10 Syndrome veinolobaire compliqué d’hypertension artérielle pulmonaire chez une femme de 70 ans ayant présenté une détresse respiratoire aiguë. Thorax standard. En radiographie standard, on retrouve, outre les signes du poumon droit hypogénétique, une image vasculaire en forme de « sabre turc » s’élargissant de haut en bas, visible dans environ deux tiers des cas, traduisant le retour veineux pulmonaire anormal à direction caudale. Cette image, appelée « signe du cimeterre », peut être masquée par la dextroversion de la masse cardiaque [57]. Elle peut être confondue avec une veine cave supérieure ectopique intra-parenchymateuse, une volumineuse artère systémique ou une veine pulmonaire unique se drainant vers l’atrium gauche. L’angioscanner précise les caractéristiques du retour veineux pulmonaire anormal (total ou partiel), son siège d’abouchement, la topographie, le calibre et la distribution de l’artère pulmonaire droite et des veines pulmonaires non concernées par le retour veineux pulmonaire anormal [53]. De même, la vascularisation systémique par les artères bronchiques ou par une autre artère provenant de l’aorte thoraco-abdominale est analysable par la même technique (voir fig. 4-9). La zone perfusée par ces branches donne un aspect d’hyperhémie du parenchyme pulmonaire droit concerné. On analysera précisément le calibre du vaisseau anormal et on recherchera des signes d’athérome, de thrombose murale ou un anévrisme, sources possibles de complications mettant en jeu le pronostic vital. Il associe les éléments du syndrome veinolobaire pulmonaire congénital à un passage d’une partie du parenchyme du lobe inférieur droit, au-delà de la ligne médiane, au contact ou plus rarement fusionné au lobe inférieur gauche [16]. Généralement, ce croisement s’effectue derrière le cœur et devant l’aorte et l’œsophage. La séparation est fréquemment matérialisée par une image scissurale qui se projette en radiographie standard à gauche de la ligne médiane et du rachis thoracique (fig. 4-11) [19]. La vascularisation de cet isthme parenchymateux est assurée par des branches artérielles pulmonaires du lobe inférieur droit. Le retour veineux pulmonaire anormal droit n’est absent que dans 20 % des cas. Le scanner hélicoïdal monocoupe, multicoupes ou à focalisation électronique démontre la discontinuité médiastinale derrière la masse cardiaque et les modalités de contact des deux lobes inférieurs [23, 27, 74] Le poumon en fer à cheval s’inscrit fréquemment dans un contexte malformatif complexe [33, 76]. Fig. 4-11 Syndrome veinolobaire et poumon en fer à cheval. Cette entité, également dénommée « duplication diaphragmatique », est parfois associée au poumon droit hypogénétique ou au syndrome veinolobaire pulmonaire congénital [53]. Dans sa genèse, on incrimine une perturbation de la descente du septum transversum (futur diaphragme) qui cloisonne la cavité cœlomique en cavités pleurales et cavité péritonéale. Une partie du septum transversum peut rester ancrée à la paroi thoracique postérieure et produire un diaphragme accessoire incomplet en forme de repli semi-lunaire pouvant fixer une partie du poumon en développement et former deux compartiments. Si le parenchyme enchâssé sous le diaphragme accessoire est aéré, il se crée une fine bande opaque, oblique, visible de face ou de profil sur l’image standard du thorax. Si le poumon n’est pas aéré, on note une opacité de type tissulaire à la base du poumon. Le scanner identifie ces deux compartiments et les continuités bronchovasculaires entre le parenchyme situé au-dessus du diaphragme accessoire et le parenchyme sous-jacent. L’incidence et l’importance clinique des variantes de la systématisation bronchique ne sont pas réellement connues. À partir des données endoscopiques ou bronchographiques anciennes, leur fréquence se situerait entre 1 et 12 % de la population [3]. Cette fréquence évolue car nos capacités de détection se sont améliorées grâce à la tomodensitométrie multicoupes et aux possibilités de reconstructions bi- ou tridimensionnelles. À titre d’exemple, le mode de division des bronches du lobe supérieur droit, siège de prédilection de variantes du normal, ne reproduit la trifurcation classique que dans à peine un tiers des cas. Il n’en reste pas moins vrai que si ces variantes du normal sont fréquentes, elles ont une incidence pathologique indéterminée, malgré leur incrimination dans la survenue des pneumothorax spontanés de l’adulte [6]. Il est habituel de distinguer les bronches dites « ectopiques » (ou « déplacées ») et les bronches surnuméraires. Les bronches « ectopiques » ont une origine déplacée, mais le parenchyme dont elles assurent la ventilation correspond à une partie ou à la totalité d’un territoire à systématisation normale. À l’inverse, les bronches « surnuméraires » assurent la ventilation d’un territoire parenchymateux qui vient se surajouter au territoire normal [21]. Les bronches ectopiques sont près de trois fois plus fréquentes que les bronches surnuméraires et se rencontrent chez environ 1 à 2 % des patients. Ce terme ambigu recouvre une variété d’anomalies bronchiques qui prennent leur origine de la trachée aux bronches souches et se dirigent vers un territoire lobaire supérieur droit, avec une prévalence de 0,1 à 2 %, ou gauche, avec une prévalence de 0,3 à 1 % [65]. Les mécanismes pathogéniques de ces malformations sont peu connus. On évoque un trouble de la morphogenèse tra-chéobronchique favorisant l’induction de bourgeons à partir de l’épithélium trachéal [18]. Ce phénomène de bourgeonnement n’existe pas normalement au niveau de la trachée. Il peut cependant y être induit si un fragment de mésenchyme bronchique actif est greffé dans l’épithélium trachéal. Les bronches surnuméraires découleraient d’un trouble de l’embryogenèse vers le 29e-30e jour. De ce fait, elles sont volontiers associées à des anomalies cardiaques précoces en raison de la relation temporelle avec le développement de ces structures de type dextrocardie, transposition des gros vaisseaux, ventricule droit à double issue ou agénésie de la valve pulmonaire. Les bronches déplacées ou ectopiques (32e jour), les plus fréquentes, sont en revanche associées à des malformations cardiovasculaires plus tardives : « sling » de l’artère pulmonaire gauche, veine cave supérieure gauche, ductus arteriosus. S’il s’agit d’une bronche trachéale déplacée, le territoire ventilé peut être plurisegmentaire, segmentaire, sous-segmentaire, voire au-dessous du niveau sous-segmentaire. Dans sa comparaison entre les bronches trachéales droites et gauches, Rémy et al. [65] précisent que le segment apical (S1) en entier ou en partie est le plus souvent concerné, associé, surtout du côté gauche, au segment dorsal (S3) dans 50 % des cas. À l’inverse, qu’il s’agisse d’une bronche trachéale droite ou gauche, le segment dorsal n’est que très rarement concerné isolément (2,5 %). La symptomatologie clinique varie d’un sujet à l’autre. Les bronches trachéales sont volontiers asymptomatiques, de découverte fortuite lors d’un examen radiographique standard ou tomodensitométrique, d’une endoscopie ou d’une intervention chirurgicale. Ailleurs, elles peuvent s’exprimer par des infections pulmonaires récidivantes, des troubles de ventilation ou des dilatations des bronches, conséquences possibles d’une sténose proximale. Signalons enfin la possibilité de troubles de ventilation en cas d’intubation trachéale avec gonflage du ballonnet de la canule en regard de la bronche ectopique ou de lésions traumatiques par intubation sélective. Les manifestations cliniques sont plus marquées pour les bronches trachéales gauches (fig. 4-12). Elles paraissent dépendre de plusieurs facteurs : la proximité de la bronche ectopique avec la crosse de l’artère pulmonaire gauche, qui pourrait agir comme élément compressif, l’existence d’une sténose congénitale et la présence d’anomalies cartilagineuses. Ces facteurs contribueraient à l’apparition de phénomènes d’hyperaération avec piégeage, d’une dilatation des bronches, d’une hypoplasie kystique, de troubles de ventilation, d’hémoptysies, voire de lymphangiectasies congénitales. Fig. 4-12 Bronche trachéale gauche. La tomodensitométrie hélicoïdale retrouve la lumière de la bronche ectopique ou surnuméraire, ainsi que son caractère normal, sténosé ou ectasique (fig. 4-13) [5]. La distribution respective des bronches orthotopiques du lobe supérieur et de la bronche ectopique ou surnuméraire est précisée, ainsi que les éventuelles complications telles que l’emphysème obstructif et les dilatations des bronches avec possible piégeage expiratoire [36]. Fig. 4-13 Bronche trachéale droite déplacée. C’est une bronche surnuméraire rare maisbien connue sur les plans anatomique, bronchographique et endoscopique. Sa fréquence se situe entre 0,09 et 0,5 %. Une prédominance masculine ressort des données de la littérature. Elle prend son origine le long de la paroi médiale de la bronche intermédiaire droite juste avant la naissance de la bronche apicale du lobe inférieur (B6) et en général en amont de la bronche lobaire moyenne (B4-B5) [5]. Des variantes sont possibles (fig. 4-14). Fig. 4-14 Bronche cardiaque accessoire. Sur le plan clinique, elle peut être découverte de façon fortuite ou à l’occasion d’une toux chronique, d’épisodes récurrents d’infection bronchopulmonaire, de dyspnée ou de suppuration chronique dans le lobe inférieur droit. Une description tomodensitométrique sur six cas a été rapportée décrivant, outre la lumière bronchique dont le diamètre oscille entre 7 et 9 mm, un éperon bronchique à son origine, une scissure séparant le parenchyme aéré du territoire de cette bronche accessoire de celui du lobe inférieur droit dans deux tiers des cas [47]. De plus, l’injection de produit de contraste apparaît précieuse pour démontrer d’une part le rehaussement de la paroi bronchique, d’autre part une prise de contraste par le parenchyme dépendant de cette bronche, situé dans le récessus inter-azygo-œsophagien. Cette prise de contraste peut poser des problèmes diagnostiques en particulier avec une adénomégalie sous-carénaire. L’endoscopie révèle la présence d’une muqueuse bronchique et l’étude anatomopathologique la présence de cartilage, ce qui permet la distinction avec les fistules ou les diverticules. Sur les 14 observations rapportées par Ghaye et al. [21], 10 étaient des culs-de-sac borgnes. Les autres débouchaient sur une masse tissulaire plus ou moins aérée localisée dans le récessus inter-azygo-œsphagien. La résection chirurgicale est indiquée chez le patient symptomatique. Nous ne ferons que citer certaines malformations à révélation pédiatrique pratiquement exclusive. – Les malformations bronchopulmonaires communicantes de l’intestin primitif (poumon œsophagien), dont 4 cas ont été rapportés par Leithiser et al. [43] : dans trois de ces observations, une bronche droite provenait de la partie basse de l’œsophage près du cardia. – La bridging bronchus (littéralement « bronche en pont ») : il s’agit d’une anomalie exceptionnelle au cours de laquelle une bronche ectopique destinée au lobe inférieur droit prend son origine dans la bronche souche gauche traversant le médiastin dans la région rétrocardiaque. Une description tomodensitométrique récente en a été faite [44]. S’intégrant généralement dans un syndrome polymalformatif, la bridging bronchus a un pronostic souvent péjoratif et conduit à des détresses respiratoires aiguës néonatales. – L’emphysème lobaire congénital, dont le caractère héréditaire est envisagé, se manifeste généralement avant le 6e mois [36, 44, 68]. Un mécanisme de valve incomplète serait à l’origine de la distension et du trappage d’un secteur parenchymateux où la constatation d’un emphysème réel sur le plan microscopique est exceptionnelle. Deux hypothèses sont envisagées : une anomalie bronchique intrinsèque au niveau du cartilage ou une multiplication anormale des alvéoles sans anomalie des voies aériennes (« emphysème polyalvéolaire lobaire »). L’emphysème lobaire congénital atteint par fréquence décroissante le lobe supérieur gauche, le lobe moyen et le lobe supérieur droit. Les signes cliniques sont classiques : dyspnée, poly-pnée, toux, wheezing, détresse respiratoire aiguë. La radiographie standard révèle une hyperclarté plus ou moins étendue accompagnée de distension, une compression du parenchyme sain et un déplacement médiastinal. La découverte à l’âge adulte est très rare. Cependant, au traitement chirurgical traditionnel vient se substituer un traitement conservateur chez les enfants peu symptomatiques [35]. L’amélioration progressive du rapport ventilation/perfusion, surtout au profit de la ventilation, et la réduction de la compression des lobes adjacents conduisent certains praticiens à préférer cette option non chirurgicale, ce qui augmentera, de facto, la fréquence de l’emphysème lobaire congénital de l’adulte. Signalons enfin le déplacement ou la fusion des bronches lobaires supérieures droites et lobaires moyennes, simulant un lévo-isomérisme [62]. Un dextro-isomérisme correspond à deux poumons trilobés et une disposition épartérielle bilatérale. La prédominance masculine est nette. Si la rate est absente, Landing [42] attribue le terme de « syndrome d’Ivemark » dans leur classification (type I). Si les rates sont multiples et de tailles inégales, il s’agit d’un type II, ou anisosplénie masculine. Dans le lévo-isomérisme, les poumons sont bilobés avec une disposition bronchique hypartérielle bilatérale (fig. 4-15). En cas de polysplénie, il s’agit d’un type III sans prédominance masculine ou féminine. Le type IV correspond à un lévo-isomérisme avec anisosplénie féminine. Fig. 4-15 Lévo-isomérisme. Angioscanographie hélicoïdale axiale. Les anomalies d’isomérisme s’intègrent volontiers dans des contextes malformatifs cardiovasculaires et abdominaux de mauvais pronostic. L’acquisition tomodensitométrique volumique fait le diagnostic et contribue à l’évaluation des malformations viscérales ou vasculaires associées. Il s’agit d’une solution de continuité complète ou incomplète d’une bronche proximale ventilant un secteur lobaire et, plus fréquemment, segmentaire, voire sous-segmentaire [54]. Dans la genèse de cette affection, on incrimine soit le détachement d’un bourgeon bronchique proximal qui poursuit sa croissance périphérique indépendamment de sa connexion proximale, soit les conséquences d’une ischémie du bourgeon bronchique survenant in utero avec involution secondaire [75]. Dans l’une ou l’autre des hypothèses, le bourgeon bronchique périphérique poursuit sa croissance au sein du mésenchyme et les espaces aériens périphériques sont primitivement normaux. Au fil du temps, la lumière bronchique en amont de la zone atrétique se remplit de sécrétions muqueuses provenant des glandes bronchiques. Il se constitue une mucocèle dont l’extension digitiforme en distalité est plus ou moins étendue. Le secteur pulmonaire concerné est aéré grâce à la ventilation collatérale assurée par les canaux bronchiolo-alvéolaires de Lambert et les pores interalvéolaires de Kohn. Aussi, en raison de l’obstruction bronchique proximale, se développent une hyperinflation et un trappage du parenchyme concerné avec compression des secteurs parenchymateux adjacents et hypovascularisation artérielle pulmonaire du secteur en distension. Le nombre de cas rapportés dans la littérature est très limité [55]. La localisation habituelle est le lobe supérieur gauche dans 64 % des cas, le lobe inférieur gauche dans 14 % des cas et le lobe moyen et le lobe inférieur droit dans 8 % des cas. Au sein du lobe supérieur gauche, B1 et B3 sont les plus fréquemment concernées. En radiographie standard et en tomodensitométrie, la bronchocèle se traduit par une masse parahilaire avec d’éventuels prolongements digitiformes périphériques tubulés en V ou en Y. Elle peut être soit pleine, soit le siège de niveaux hydroaériques ou d’air [84]. Plus rarement, les mucosités au sein de la mucocèle peuvent être calcifiées, voire mobiles, en fonction de la position. Lorsqu’elle est pleine, la bronchocèle est de densité homogène et ne se rehausse pas après injection de produit de contraste. L’association bronchocèle et hyperinflation est pratiquement constante dans l’atrésie bronchique congénitale mais n’est en rien spécifique de cette dernière puisqu’on la rencontre également en cas de masse compressive hilaire proximale (tumeur bénigne ou maligne, kyste bronchogénique pédiculaire compressif, corps étrangers, broncholithiase) (fig. 4-16). Le diagnostic différentiel est particulièrement délicat en regard du hile gauche ; il comprend, outre le kyste bronchogénique, l’emphysème lobaire congénital ou une bronche trachéale [65]. Là encore, le rôle du scanner à acquisition volumique avec reconstructions multiplanaires et 3D est majeur pour éliminer un élément compressif proximal, préciser la topographie du segment bronchique atrétique et rechercher des malformations associées (séquestration, retour veineux pulmonaire anormal, veine cave supérieure gauche persistante, etc.) [5]. Fig. 4-16 Atrésie bronchique du segment de Fowler droit. Sur les 9 cas rapportés par Matsuchima et al. [55], l’association radiologie-endoscopie a fait le diagnostic sept fois. Deux fois il s’agissait d’un diagnostic opératoire ; en effet, l’hyper-aération n’existait pas au scanner car le secteur parenchymateux concerné était de petite taille. Le rôle de l’imagerie par résonance magnétique est encore imprécis. Elle serait utile pour la détection des impactions qui se traduisent généralement par un haut signal en T1 et T2 [77]. À l’inverse, l’exérèse chirurgicale s’impose en cas de surinfection et de suppuration. L’analyse précise de la systématisation du secteur concerné par le scanner volumique est essentielle avant l’étape chirurgicale [64]. Elle est complexe en raison des redistributions de parenchyme, secondaires à l’hyperaération. En théorie, une exérèse chirurgicale la plus limitée possible est préférable. Cependant, la fréquence des fuites aériques postopératoires dans les résections atypiques est supérieure à celles constatées lors des lobectomies. Ces anomalies résultent du développement anormal d’un bourgeon bronchique détaché de la partie trachéobronchique de l’intestin primitif au cours de sa différenciation in utero [77]. Il conduit à la formation d’une poche de liquide généralement sans issue qui reste satellite de la trachée, des bronches ou de l’œsophage, avec lesquels une communication peut exister (fig. 4-17). Si le détachement est précoce, le dérivé kystique pourra garder un contact médiastinal, proximal s’il se situe au-dessus du clivage diaphragmatique (septum transversum), ou caudal avec de possibles localisations sous-diaphragmatiques au contact ou au sein des viscères abdominaux, voire à cheval de part et d’autre du diaphragme [11] (fig. 4-18). Beaucoup plus rarement, des ectopies non communicantes peuvent exister à distance des structures médiastinales ou digestives (creux sus-claviculaire, région rétrosternale, territoire sous-cutané, etc.) [1]. Si le détachement est plus tardif lors de l’embryogenèse, il donne lieu à des localisations pulmonaires [29, 48, 71]. Fig. 4-17 Kyste bronchogénique intramural communicant de l’œsophage. La radiographie standard du thorax de face montrait une opacité médiastinale en projection cardiaque. Fig. 4-18 Kyste bronchogénique thoraco-abdominal en « sablier ». Macroscopiquement, les kystes bronchogéniques sont des poches à contenu liquidien dans tous les cas, sphériques ou elliptiques, dont le diamètre moyen se situe aux alentours de 5 cm. Le contenu est laiteux, séreux, gélatineux ou épais, blanc et translucide ou, au contraire, verdâtre, brun, jaune ou à contenu purulent ou hémorragique. Deux des 68 cas rapportés par Mac Adams et al. [46] avaient un contenu gazeux. La poche ne présente pas de cloisons internes. Les kystes bronchogéniques sont plus volontiers uniques et uni-loculaires. Microscopiquement, les kystes sont revêtus d’un épithélium respiratoire pseudo-stratihé ou en colonne. Leur paroi contient des glandes, du cartilage, ainsi que des éléments musculaires lisses. Pour certains auteurs, la présence de cartilage est un élément déterminant du diagnostic différentiel avec les autres formations kystiques congénitales [46]. La localisation médiastinale des kystes bronchogéniques est prépondérante (75 à 80 %). Les kystes bronchogéniques représentent 10 à 15 % des masses primitives du médiastin. Soixante-dix-neuf pour cent sont localisés dans le médiastin moyen, 17% dans le médiastin postérieur et 3 % dans le médiastin antérieur, incluant les localisations intrapéri-cardiques [46]. En raison de leur mécanisme de formation, la majorité d’entre eux ont une topographie paratrachéale, autour de la carène, en arrière de la région hilaire, au contact ou à l’intérieur de la paroi œsophagienne, dans le ligament pulmonaire ou les gouttières costovertébrales [2]. Ils ont tendance à se mouler sur les structures adjacentes et à sortir de la région de la bifurcation trachéobronchique vers l’arrière et la droite en avant de la colonne vertébrale. Pour ce qui concerne les kystes pédiculaires gauches, les possibilités de croissance sont beaucoup plus limitées par la présence de la crosse de l’aorte et du ligament artériel, ce qui explique la fréquence des complications compressives des éléments hilaires. L’histoire naturelle des kystes bronchogéniques est difficile à établir puisque les séries de suivi longitudinal chez des patients non opérés sont peu nombreuses. La fréquence des patients symptomatiques s’échelonne entre 56 et 67 % [1, 46, 58, 67, 71]. Certains symptômes sont progressifs : douleur, toux, dyspnée, wheezing, dysphagie. En cas de croissance progressive, ou rapide à l’occasion d’une complication (fistulisation, infection, hémorragie), ils peuvent provoquer des compressions cardiaques et vasculaires (fig. 4-19). Fig. 4-19 Kyste bronchogénique médiastinal surinfecté compliqué de syndrome cave supérieur aigu.
EMBRYOLOGIE
Développement pulmonaire
Développement intra-utérin
Développement vasculaire
Vaisseaux pulmonaires périphériques
Circulation bronchique
POUMON DYSMORPHIQUE
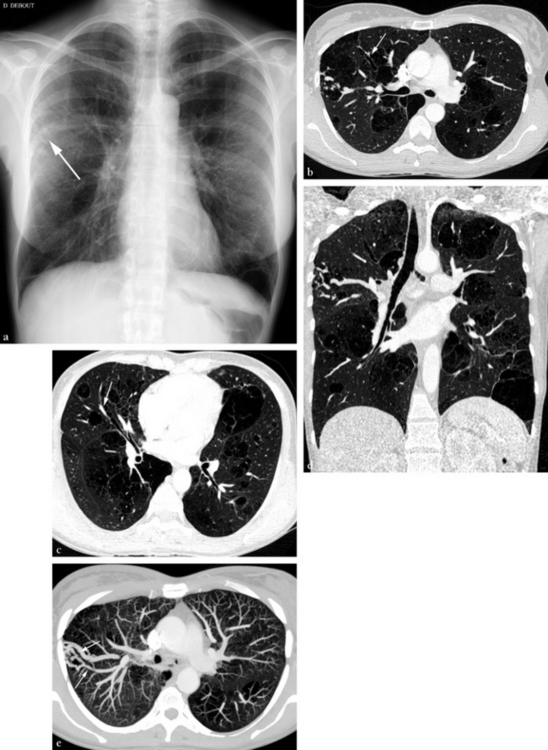
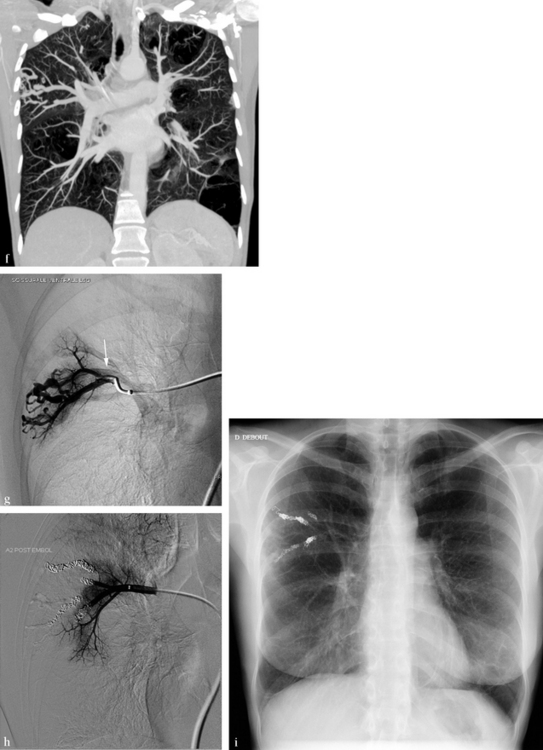
Il s’agit d’une femme de 46 ans ayant présenté une amaurose gauche. Dans ses antécédents, on signale la découverte à 35 ans d’une pathologie bronchopulmonaire malformative à l’occasion d’une pneumopathie du lobe moyen. (a) Radiographie standard permettant de déceler des images bulleuses bilatérales et des opacités serpigineuses en région axillaire droite (flèche). (b) Reformation axiale transverse d’un angioscanner pulmonaire. Mise en évidence d’images bulleuses circonscrites associées à des dilatations des bronches, visibles ici au niveau de B2 droite (flèche). En région axillaire droite, des vaisseaux dilatés siègent au sein d’une zone bulleuse. (c) Reformation axiale oblique dans le plan de la bronche lobaire moyenne. Aspect de dilatation des bronches affectant B4 droite (flèche). (d) Reformation coronale oblique dans le plan de la trachée. On identifie le raccordement proximal de la malformation vasculaire axillaire droite. (e) Reformation axiale MIP centrée sur la malformation vasculaire axillaire droite. On identifie une artère afférente (flèche) provenant d’une sous-segmentaire dorsale du lobe supérieur, une communication artérioveineuse au contact de la paroi et une veine efférente de plus gros calibre que l’artère (flèche). Il s’agit donc d’une malformation artérioveineuse pulmonaire (MAVP) associée à une malformation bronchopulmonaire. L’amaurose est la conséquence d’une embolie systémique paradoxale liée au shunt droite-gauche de la MAVP. (f) Reformation coronale MIP montrant la disposition des artères alimentant la MAVP, provenant d’une scissurale dorsale du lobe supérieur droit. L’indication d’une embolisation est posée en raison du calibre des artères afférentes de la MAVP (plus de 3 mm) et du caractère symptomatique de la malformation vasculaire. Le but est de prévenir une récidive d’embolie systémique. (g) Angiographie hypersélective d’une branche alimentant une partie de la MAVP. Les rameaux alimentant la MAVP ont un calibre irrégulier et un aspect dysplasique. On visualise le retour veineux précoce de la MAVP (flèche). L’embolisation sera faite par largage de spirales non ferromagnétiques ou en platine. (h) Angiographie de contrôle en fin d’embolisation montrant une exclusion complète de la MAVP. (i) Radiographie standard pratiquée après l’embolisation, montrant les spirales d’embolisation non ferromagnétiques standard (peu opaques) ou en platine (très opaques).
Agénésie, aplasie et hypoplasie pulmonaires
Hypoplasie pulmonaire harmonieuse
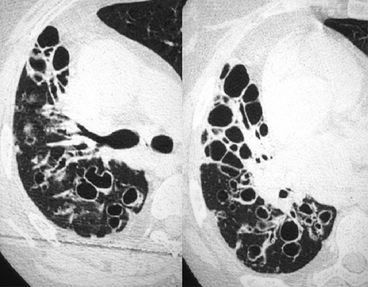
Au sein de ce poumon droit réduit de volume (déplacement cardiomédiasti-nal et hernie transmédiastinale antérieure du poumon gauche), des lésions kystiques en partie de siège sous-pleural sont associées à des bronches proximales à parois épaisses et discrètement irrégulières, Le diagnostic différentiel avec des bronchectasies kystiques est impossible en regard du segment de Fowler. Noter l’existence de petites sécrétions endocavitaires.
Agénésie, aplasie et hypoplasie lobaires
Agénésie lobaire
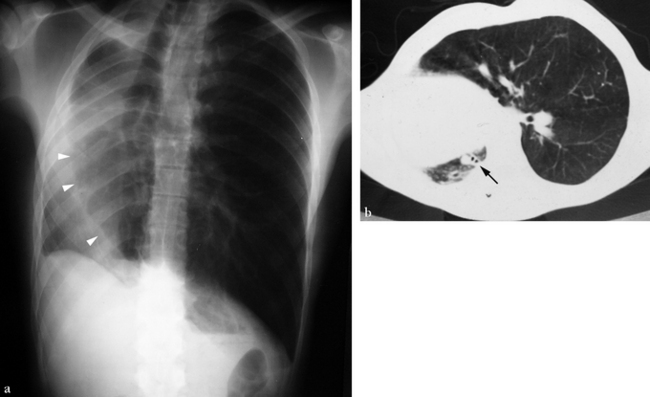
(a) Thorax standard. Il existe une rétraction hémithoracique droite majeure, un déplacement vers la droite de la masse cardiomédiastinale et une hernie trans-médiastinale antérieure du poumon gauche. La limite antérieure du poumon gauche est visible (têtes de flèche). Les lumières des bronches lobaires supérieure droite et moyenne sont invisibles. (b) La seule lumière bronchique droite (flèche) se distribue à une petite zone parenchymateuse en projection postéro-basale sur la tomodensitométrie axiale. L’aspect radiographique standard est très proche de celui d’une agénésie pulmonaire droite.
(a) Coupe au niveau de la carène. Présence d’une scissure séparant le territoire lingulaire en avant du territoire lobaire inférieur en arrière (flèche). Les lumières bronchiques situées en dehors de l’artère pulmonaire gauche correspondent à des bronches lingulaires. Présence d’une veine cave supérieure gauche se drainant vers l’atrium gauche et d’une crosse aortique droite. Petit déplacement médiastinal vers la gauche. (b) Coupe en regard de la zone de naissance de la bronche lobaire supérieure gauche. Aucune bronche culminale n’est visible. (c) Coupe à un niveau inférieur à celui de b montrant la bronche lingulaire et sa division (flèche).
Aplasie lobaire
(a) Tomodensitométrie axiale montrant l’interruption en cul-de-sac de la lumière de la bronche B1-B3 (flèches). La disposition bronchique proximale est de type hypartérielle. (b) Reconstruction 3D surfacique illustrant l’aspect de B1-B3 borgne et la normalité de B2 et des bronches lingulaires.
Hypoplasie lobaire
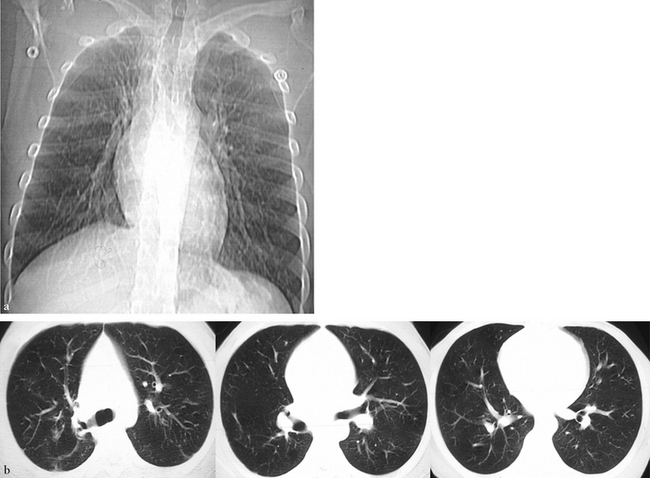
Aspect radiographique de face (a) et coupes TDM en fenêtre parenchymateuse pulmonaire dans un ordre crânio-caudal (b). Le poumon droit est de petite taille mais de morphologie harmonieuse. On constate une surélévation du hile droit et une diminution de volume du lobe supérieur droit. Les bronches sont toutes présentes mais de petite taille en calibre et en longueur (b).
Syndrome du poumon hypogénétique
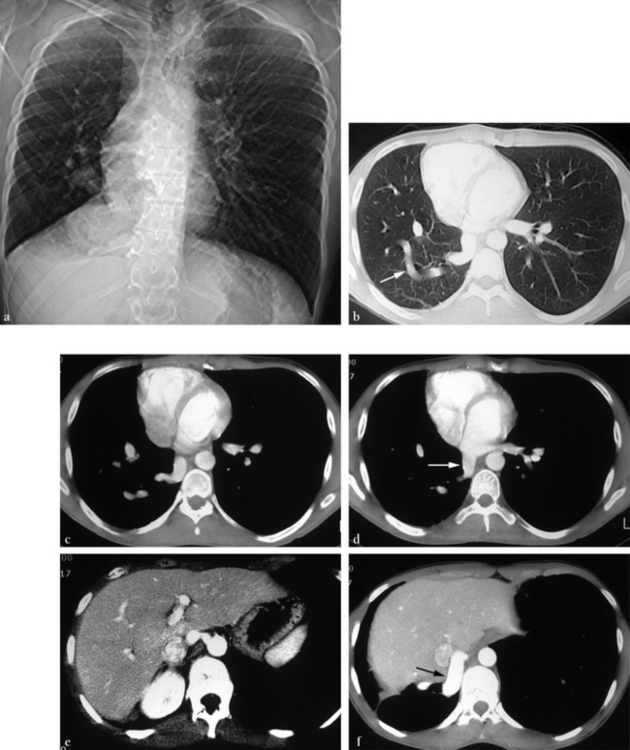
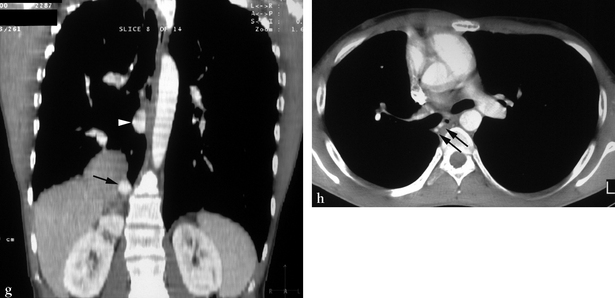
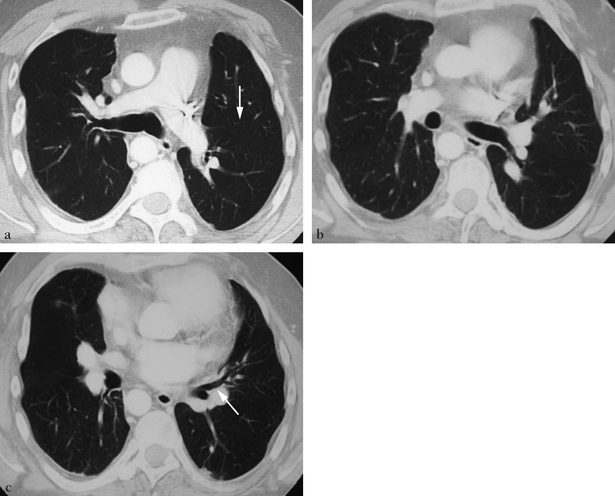
(a) Thorax de face (scout view) montrant un déplacement à droite de la silhouette cardiomédiastinale, une surélévation de la partie interne de la coupole diaphragmatique et des vaisseaux pulmonaires droits dont la répartition est anormale. (b) Tomodensitométrie en fenêtre parenchymateuse. Le vaisseau anormal décrivant une courbe (flèche) correspond à une branche supérieure d’une veine pulmonaire se dirigeant vers le bas. (c, d) Coupes sous-jacentes à b montrant le collecteur veineux unique se drainant normalement vers l’atrium gauche, après un trajet anormal (flèche). (e) Coupe abdominale. Une artère anormale naît du tronc cœliaque. (f) Cette artère systémique est volumineuse, se dirigeant vers la région pulmonaire postéro-basale (flèche). (g) Reconstruction coronale montrant la hernie diaphragmatique, l’artère systémique (flèche) et l’abouchement du collecteur veineux dans l’atrium gauche (tête de flèche). (h) Fenêtre médiastinale en projection hilaire montrant des artères bronchiques de gros calibre (flèches). Source : clichés Jean Beaugrand, scanner Corbeil.
Syndrome veinolobaire pulmonaire congénital ou syndrome du cimeterre [40]
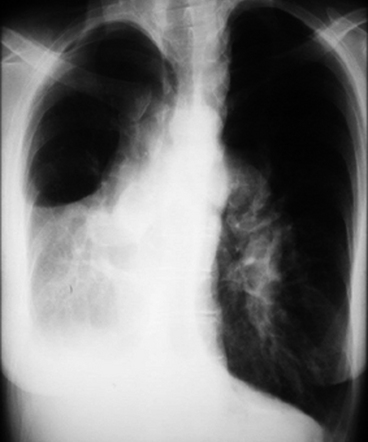
Aspect d’hypoplasie pulmonaire droite avec rotation cardiomédiastinale. L’image du cimeterre n’est pas visible. Dilatation majeure bilatérale des artères pulmonaires traduisant l’hypertension artérielle pulmonaire, conséquence tardive d’un shunt gauche-droite significatif non corrigé.
Poumon dit en « fer à cheval »
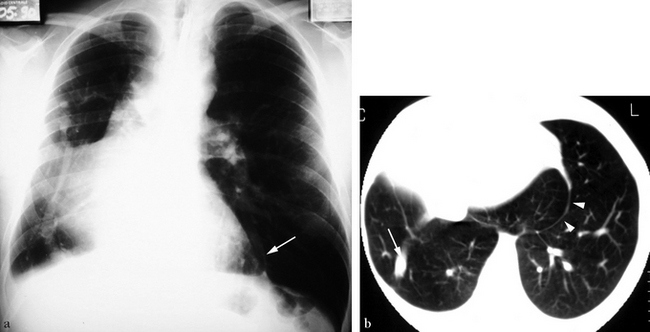
(a) Thorax standard. Syndrome du cimeterre avec hypoplasie droite et déplacement cardiomédiastinal. Une image scissurale en projection basale gauche traduit le contact des deux segments postéro-basaux des lobes inférieurs droit et gauche (flèche). (b) Tomodensitométrie en fenêtre pulmonaire. Visibilité du retour veineux pulmonaire anormal à la base droite (flèche). L’isthme parenchymateux transmédiastinal droit appartient au segment postéro-basal et vient au contact du lobe inférieur gauche, créant l’image scissurale (têtes de flèche).
Diaphragme accessoire
ANOMALIES TRACHÉO-BRONCHO-ŒSOPHAGIENNES
Anomalies de division bronchique [15]
Bronches « trachéales »
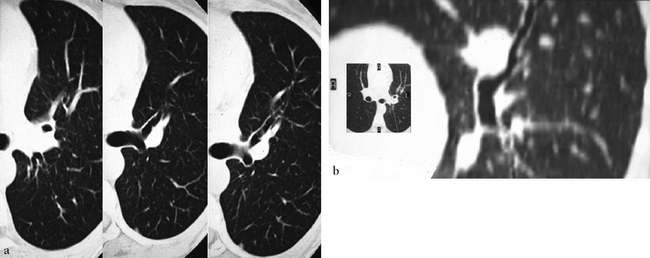
Coupe de 3 mm en fenêtre parenchymateuse pulmonaire. (a) Une bronche en forme de cul-de-sac naît de la bronche lobaire supérieure gauche. (b) D’orientation oblique en dehors et en arrière, elle est surplombée par la crosse de l’artère pulmonaire gauche. Sur la reformation, la bronche antérieure est à destinée des territoires culminal restant et lingulaire.

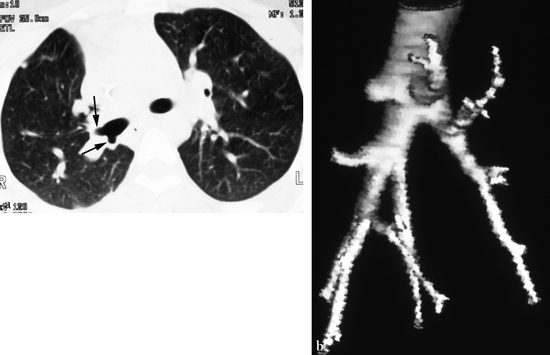
(a) Tomodensitométrie axiale en fenêtre parenchymateuse montrant une clarté bronchique naissant de la face latérale droite de la trachée. (b) Reformation coronale illustrant le trajet d’abord descendant de la bronche ectopique, puis ascendant après passage sous la crosse de la veine azygos. Le territoire parenchymateux correspond au segment 1 droit. Les bronches destinées aux segments 2 et 3 étaient normales. (c) Endoscopie virtuelle montrant l’éperon entre la lumière trachéale et celle de la bronche ectopique.
Bronche cardiaque accessoire [80]
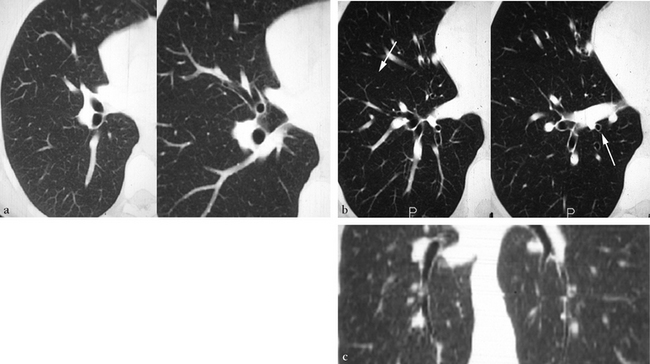
(a, b) Coupes TDM de 3 mm d’épaisseur en fenêtre parenchymateuse pulmonaire dans un ordre crânio-caudal. Une clarté annulaire naît en regard de la face inférieure de la bronche lobaire moyenne juste avant sa division en branches segmentaires interne et externe, aérant une portion du territoire paracardiaque à travers une grande scissure incomplète. La segmentaire paracardiaque est quant à elle de topographie rétroveineuse pulmonaire inférieure (flèche). (c) Une reformation coronale oblique objective la direction caudale de cette bronche surnuméraire.
Autres malformations
Anomalies d’isomérisme
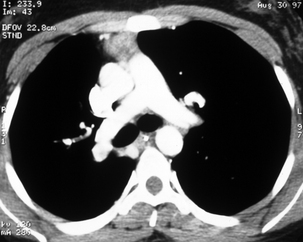
La disposition à droite est du type bronche hypartérielle caractéristique d’un lévo-isomérisme.
Atrésies bronchiques congénitales
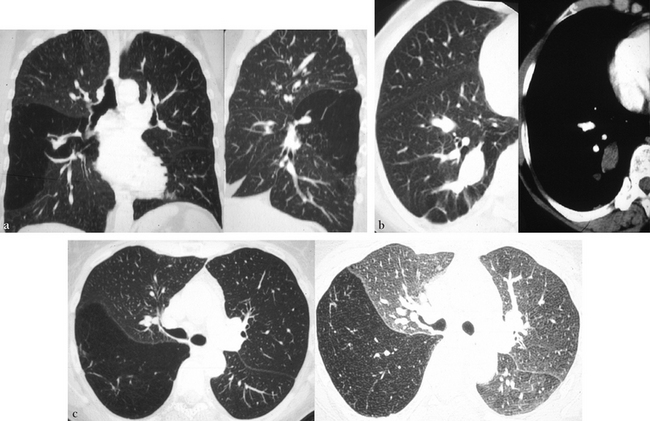
(a) Reformation coronale et sagittale à partir d’une acquisition volumique en coupes de 5 mm reconstruites tous les 3 mm. Mise en évidence d’une hyperaération de S6. (b) Présence d’une formation oblongue, pleine, non rehaussée, dont l’orientation antéro-postérieure évoque une bronchocèle du segment de Fowler droit. Noter l’absence de la bronche segmentaire normale à ce niveau. (c) Hyperaération manifeste en inspiration (image de gauche) et piégeage en expiration (image de droite).
Dérivés kystiques de l’intestin primitif
Dérivés kystiques de l’intestin primitif antérieur
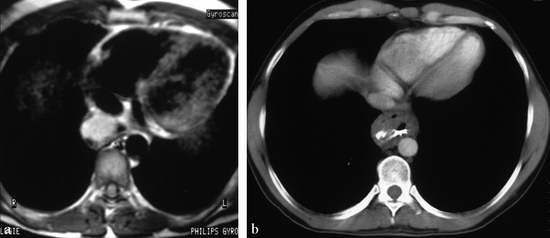
(a) IRM en SE T1. Masse en hypersignal adjacente à l’œsophage se projetant en avant de l’azygos et à droite de l’aorte thoracique descendante. (b) Tomodensitométrie axiale avec ingestion et injection de produits de contraste. La masse tissulaire fait corps avec l’œsophage dont la paroi est épaissie. Des bulles gazeuses occupent la partie antérieure de la masse. Passage du contraste ingéré de la lumière œsophagienne vers la partie latérale du kyste.
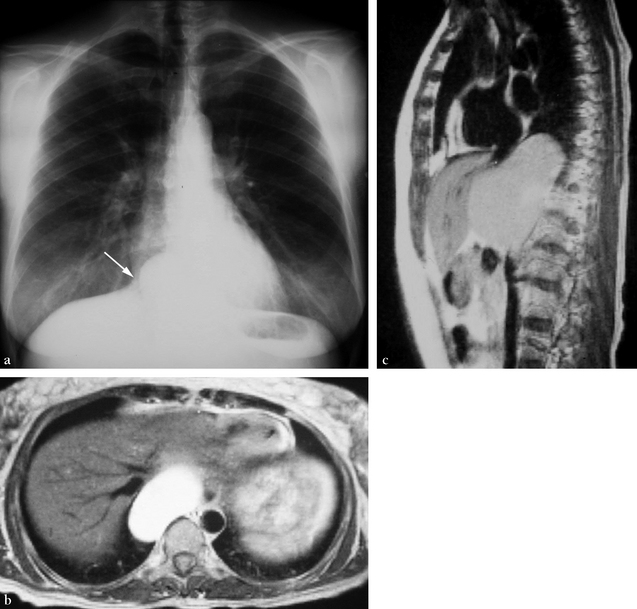
(a) Thorax standard. Opacité arrondie médiastinale en projection cardiaque droite basse dont le contour externe s’efface en regard du diaphragme, évoquant un passage transdiaphragmatique (signe de l’iceberg) (flèche). (b) IRM axiale SE T1. Masse ovoïde médiastinale en hypersignal franc homogène s’étendant depuis le secteur postéro-basal droit jusqu’au-delà de la ligne médiane au contact de l’aorte descendante. Refoulement vers l’avant du foie en regard de l’abouchement des veines hépatiques. (c) IRM sagittale latéralisée à droite de la ligne médiane en SE T1. La masse piriforme à renflement inférieur sous-diaphragmatique refoule vers l’avant le foie et, en sus-diaphragmatique, la veine cave inférieure juxtacardiaque.
Kystes bronchogéniques médiastinaux
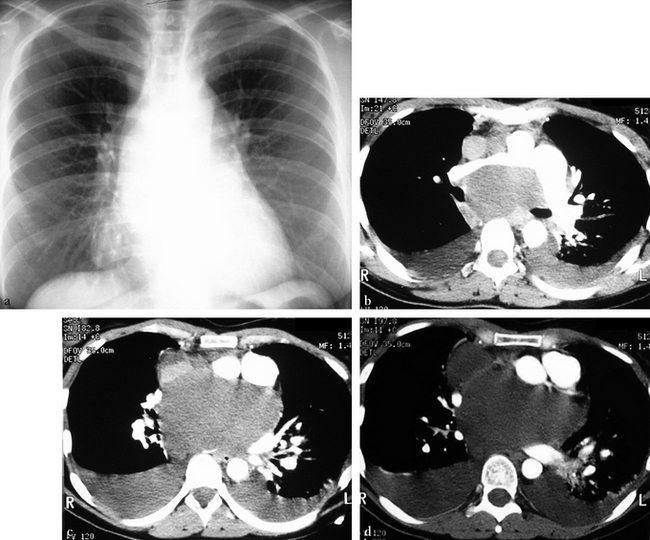
(a) Thorax standard montrant un élargissement médiastinal important avec refoulement latéral de la bronche intermédiaire droite, évoquant une masse sous-carénaire. (b) Angioscanographie hélicoïdale axiale avec injection bipédieuse, niveau sous-carénaire. Volumineuse masse kystique homogène de densité un peu supérieure à celle des épanchements pleuraux liquidiens. On note un refoulement et un étirement du tronc de l’artère pulmonaire droite. La veine cave supérieure non opacifiée est augmentée de calibre. (c) Angioscanographie hélicoïdale axiale. Il existe un refoulement important vers l’avant de l’artère pulmonaire, de l’aorte et de la veine cave supérieure. Les éléments hilaires droits sont refoulés latéralement et les veines pulmonaires gauches sont laminées. (d) Angioscanographie hélicoïdale axiale. La veine cave supérieure n’est plus visible.![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree








