141 Glomérulonéphrite
Introduction
Le terme « glomérulonéphrite » désigne une lésion inflammatoire des glomérules qui se traduit par une hématurie, une protéinurie, de l’œdème, de l’hypertension et de l’urémie. Parmi les nombreuses maladies glomérulaires, chacune caractérisée par ses caractéristiques pathologiques propres, son évolution naturelle et sa réponse au traitement, beaucoup se manifestent par un syndrome clinique semblable. Par exemple, une hématurie microscopique asymptomatique est une conséquence de plusieurs entités pathologiques disparates, le plus souvent une néphropathie à membrane basale mince et une néphropathie à l’immunoglobuline A (IgA). Ainsi, lors de l’évaluation des patients atteints de glomérulonéphrite, il est utile de déterminer d’abord à quelle catégorie clinique générale ils appartiennent, puis de distinguer les différentes maladies au sein de chaque groupe, comme l’encadré 141.1 le décrit.
Étiologie et pathogénie
Plusieurs mécanismes peuvent aboutir à une glomérulonéphrite. Le plus commun est une lésion inflammatoire qui résulte de la formation de complexes immuns et leurs dépôts dans la membrane basale glomérulaire ou le mésangium. Ces complexes immuns déclenchent plusieurs processus inflammatoires, en particulier l’activation du complément ainsi que la production et la libération de cytokines et de chimiokines. En conséquence, des cellules inflammatoires circulantes, notamment des neutrophiles, des monocytes et des lymphocytes, infiltrent les glomérules et les compartiments tubulo-interstitiels environnants. Les dépôts glomérulaires peuvent se former de trois manières :
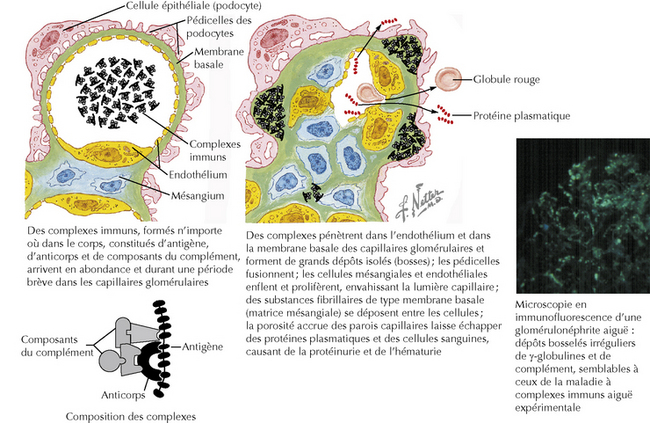
Figure 141.1 Pathogénie hypothétique des lésions glomérulaires aiguës par des complexes immuns circulants (schéma).
Les néphrites héréditaires, comme le syndrome d’Alport et la maladie à membrane basale mince, sont liées à des mutations dans le collagène de type IV. Dans le syndrome d’Alport, les mutations touchent soit la chaîne α5 du collagène de type IV (transmission liée à l’X), soit les chaînes α3 ou α4 (transmission autosomique récessive ou dominante). La maladie à membrane basale mince, une affection autosomique dominante relativement fréquente, est également liée à des mutations des chaînes α3 ou α4. Bien que les deux syndromes se présentent avec une hématurie glomérulaire asymptomatique, ils se distinguent par leur évolution naturelle. La maladie à membrane basale fine (aussi appelée « hématurie familiale bénigne ») a un pronostic favorable à long terme, alors que le syndrome d’Alport entraîne une perte progressive de la fonction rénale. Certains parents atteints du syndrome d’Alport peuvent également être atteints de surdité neurosensorielle, d’anomalies oculaires, de retard mental ou de léiomyomatose.
Tableau clinique
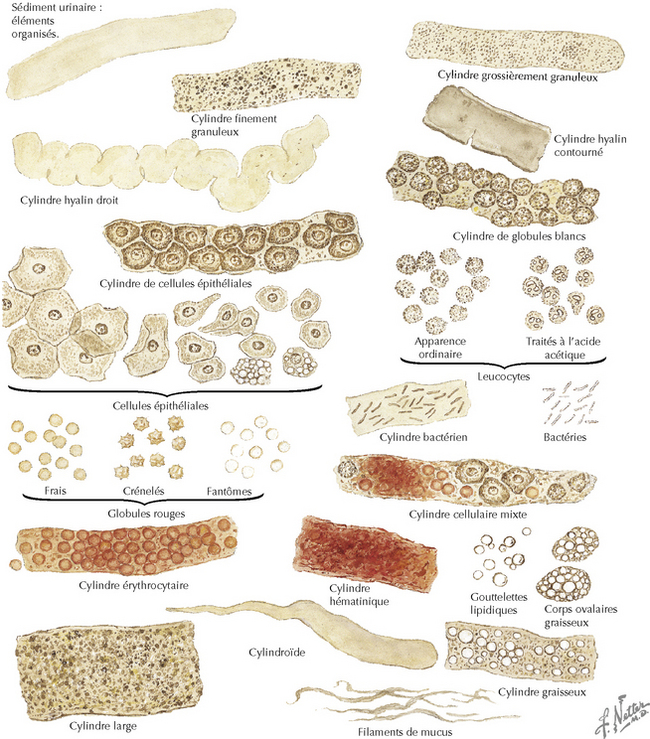
L’expression clinique de la maladie est en corrélation avec les caractéristiques pathologiques ; plus agressive est la lésion pathologique, plus grave est la maladie. Certains patients ont une hématurie asymptomatique et une hématurie macroscopique récurrente. L’hématurie, définie sur la base de la présence de plus de trois globules rouges par champ dans le sédiment urinaire obtenu par centrifugation et examiné au fort grossissement, peut être d’origine glomérulaire ou non glomérulaire (figure 141.2). En passant par le néphron, les globules rouges sont déformés (acanthocytes) sous l’effet de la composition chimique de l’environnement. Ces acanthocytes sont quasi pathognomoniques de saignement glomérulaire. Cette anomalie associée à des cylindres de globules rouges, à d’autres types de cylindrurie et à de la protéinurie suggère que l’hématurie est d’origine glomérulaire et ne provient pas du tractus urinaire.
Les causes les plus communes d’hématurie asymptomatiques sont énumérées dans l’encadré 141.1. La néphropathie à IgA peut se manifester sous la forme de trois syndromes différents. Environ la moitié de ces patients ont des antécédents d’épisodes d’hématurie macroscopique associés à une infection des voies respiratoires supérieures qui peuvent se reproduire, lors d’épisodes ultérieurs de pharyngite, d’une maladie fébrile ou d’un gros effort. Les patients sont généralement asymptomatiques ; l’hypertension artérielle et l’œdème périphérique sont rares. Le deuxième syndrome le plus courant se caractérise par une hématurie microscopique asymptomatique avec protéinurie ; à long terme, il a le plus mauvais pronostic. Le troisième type s’inscrit dans le cadre du syndrome de vasculite, le purpura rhumatoïde (Henoch-Schönlein), qui se développe le plus souvent chez les enfants. Le syndrome néphrétique postinfectieux de la néphropathie à IgA se distingue de la glomérulonéphrite poststreptococcique en ce qu’il survient 1 à 2 j après le début de l’infection, alors que la glomérulonéphrite poststreptococcique se manifeste 10 à 14 j après l’infection.
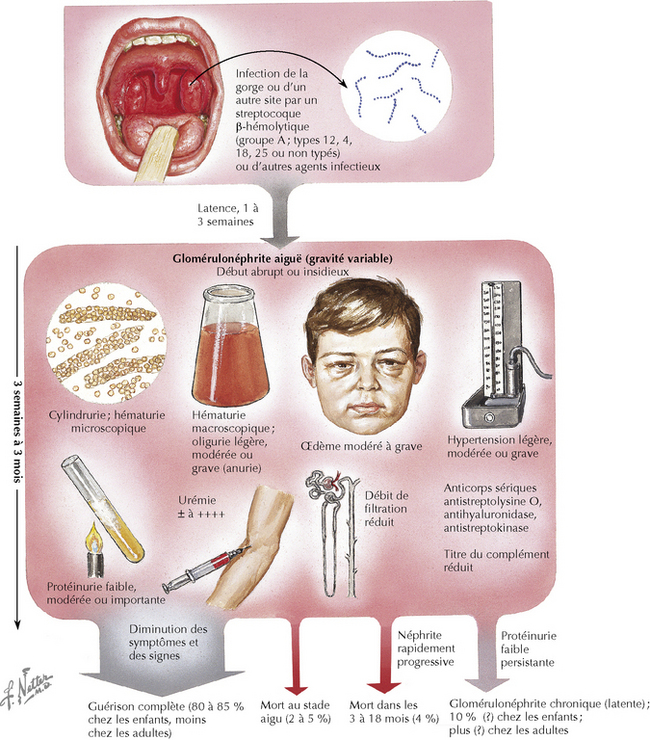
La glomérulonéphrite aiguë se manifeste habituellement par l’apparition soudaine d’une protéinurie, d’une hématurie et de sédiments urinaires contenant des globules rouges dysmorphiques, des cylindres érythrocytaires et des débris cellulaires. Les patients ont généralement de l’œdème, de l’hypertension, une oligurie et une insuffisance rénale. Dans la forme la plus sévère, la glomérulonéphrite rapidement progressive (GNRP), la concentration de créatinine sérique s’élève en quelques jours ou semaines, entraînant une insuffisance rénale grave. Structurellement, la GNRP est généralement associée à la formation de croissants glomérulaires qui résultent de la prolifération des cellules épithéliales glomérulaires et de phagocytes mononucléaires dans l’espace de Bowman. Les croissants ne sont pas révélateurs d’une cause spécifique de lésions glomérulaires, mais peuvent relever de nombreux mécanismes pathogéniques différents.
Toutes les formes de néphrite aiguë, y compris la GNRP, peuvent être la conséquence d’infections streptococciques ou staphylococciques, ou d’affections immunologiques. La maladie se caractérise par l’apparition soudaine d’un œdème et d’une oligurie avec une protéinurie importante et une hématurie (figure 141.3). Hypertension hypertrophie cardiaque et œdème pulmonaire peuvent compliquer l’évolution clinique. Dans le monde occidental, les types d’infections entraînant une glomérulonéphrite postinfectieuse ont changé ; c’est ainsi que les patients immunodéprimés, parce qu’ils sont atteints de cirrhose, de cancer ou sont transplantés, développent des glomérulopathies dues à divers micro-organismes à Gram négatif.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


