8. Chimiothérapie anticancéreuse
Introduction – Généralités
On dit que la chimiothérapie est :
– adjuvante quand elle intervient après le traitement radiochirurgical pour traiter des micrométastases ou compléter une exérèse incomplète ;
– néoadjuvante quand elle survient avant le traitement locorégional du cancer. Elle permet alors de traiter des métastases (quasiment toujours présentes au moment du diagnostic) ou de réduire la masse tumorale en préopératoire ;
– palliative, quand il semble illusoire d’espérer guérir la maladie. Elle a alors pour objectif d’augmenter significativement la survie, ou au moins de permettre une survie sans que la maladie ne progresse, et éventuellement avec une amélioration de la qualité de la vie.
On peut diviser les médicaments anticancéreux en :
– agents cytotoxiques qui induisent une mortalité cellulaire par action directe ou indirecte sur le matériel génétique de la cellule (acide désoxyribonucléique [ADN] et acide ribonucléique [ARN]) ou des protéines nécessaires à la division cellulaire (figure 8.1) ;
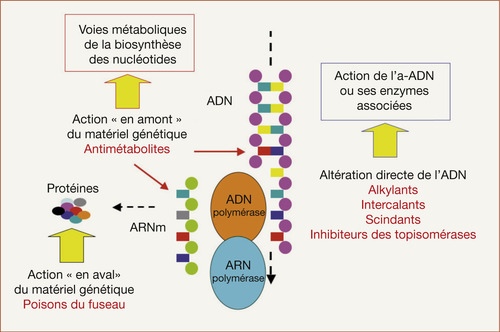
– modulateurs de la réponse biologique qui soit :
• affectent les capacités de défense de l’hôte (interleukine-2, interféron-α),
• agissent sur le contrôle hormonal de la tumeur (hormonothérapie),
• constituent les nouvelles thérapies ciblées qui contrôlent l’appareil de signalisation de la cellule (anticorps monoclonaux dirigés contre des récepteurs et bloqueurs des tyrosines kinases).
Sélectivité des médicaments anticancéreux
Les cellules cancéreuses indésirables proviennent directement de l’organisme qui les héberge et lui ressemblent donc fortement. Toute la difficulté réside dans le choix de la molécule active, à laquelle les cellules cancéreuses devront être plus sensibles que les cellules saines. De ce degré de sélectivité dépendra le niveau d’efficacité du produit au regard de ses effets indésirables et toxiques.
L’utilisation d’associations de plusieurs molécules (polychimiothérapie) permet de potentialiser les effets anticancéreux, et de diminuer les doses et donc la toxicité de chacune des molécules prises séparément (exemples dans le tableau 8.1).
| Cancer du sein | FEC 100 | Cure toutes les 3 semaines de : 5FU + épirubicine + cyclophosphamide Perfusion IV |
| TCH | Cure toutes les 3 semaines de : Docétaxel IV à J1 + Carboplatine IV à J1 + Trastuzumab IV à J0 dose de charge puis hebdomadaire | |
| Cancer colorectal | FOLFOX | Cure toutes les 2 semaines de : Oxaliplatine IV à J1 + fluoro-uracile IV + leucovorine IV |
| LV5FU | Cure toutes les 2 semaines de : Leucovorine IV à J1 et J2 + 5 fluoro-uracile IV (bolus) + perfusion continue à J1 et J2 |
La toxicité peut aussi être diminuée par l’administration locale des médicaments (administration intra-hépatique dans le traitement de tumeurs hépatiques).
Se sont développées des thérapeutiques dites « ciblées » qui ont une sélectivité d’action plus forte vis-à-vis des cellules cancéreuses. C’est ainsi que des médicaments ciblant le récepteur de l’Epidermal Growth Factor (EGFR) et son activité tyrosine-kinase sont arrivés sur le marché.
Hétérogénéité de la population cellulaire cancéreuse
Toutes les cellules d’une tumeur ne sont pas au même moment dans la même phase du cycle cellulaire, on dit qu’elles ne sont pas synchrones.
Le cycle cellulaire se compose de la succession des phases suivantes :
– la phase G1, (ou G0 pour des cellules quasiment quiescentes) est la plus longue et la plus variable ;
– la phase S suit la phase G1 Elle se caractérise par une activité intense de synthèse d’ADN en préparation à la réplication ;
– la phase G2 permet la constitution de l’appareil mitotique nécessaire à la division, ou mitose, de la cellule (polymérisation des microtubules entre autres) ;
– la mitose ou phase M est l’aboutissement de la succession des phases S et G2.
Certains médicaments sont dits « cycle-dépendants » car ils n’agissent que sur les cellules engagées dans le cycle cellulaire quelle qu’en soit la phase (exemple des agents alkylants).
D’autres produits sont « phase-dépendants », c’est-à-dire actifs pendant une phase précise du cycle (exemple des poisons du fuseau mitotique qui sont actifs en phase M).
Développement de résistances
Des cellules cancéreuses peuvent soit résister à un traitement d’emblée (résistance constitutive), soit acquérir leur résistance en cours de traitement ou lors d’une rechute alors qu’elles étaient initialement sensibles (résistance acquise). Cette acquisition de résistance peut provenir d’une pression de sélection de cellules d’emblée résistantes au sein de la tumeur initiale ou provenir de l’instabilité génétique de la tumeur qui produit des modifications de la cible des anticancéreux.
Médicaments anticancéreux : modes d’action
Anticancéreux cytotoxiques (vue générale : figure 8.1)
ACTION EN «AMONT » DU MATÉRIEL GÉNÉTIQUE : ANTIMÉTABOLITES
Ce sont :
– soit des analogues structuraux ou faux substrats des bases puriques (adénine, guanine) ou pyrimidiques (thymine, cytosine, uracile) qui vont s’incorporer dans l’ADN et l’ARN à la place de ces bases, et bloquer la transcription de l’ADN et donc de la cellule ;
– soit des inhibiteurs de la synthèse de l’acide folinique, indispensable à la synthèse des purines.
Le tableau 8.2 mentionne tous les médicaments appartenant à ce groupe.
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Antipyrimidiques | |
| Fluoro-uracile (5-FU) | Fluorouracile |
| Capécitabine | Xéloda |
| Gemcitabine | Gemcitabine, Gemzar |
| Tegafur-uracile | UFT |
| Cytarabine | Aracytine, Cytarabine, Depocyte |
| Azacitidine | Vidaza |
| Antipuriques | |
| Mercaptopurine | Purinéthol |
| Azathioprime | Imurel |
| Fludarabine | Fludara |
| Cladribine | Leustatine, Litak |
| Clofarabine | Evoltra |
| Nélarabine | Atriance |
| Pentostatine | Nipent |
| Antifoliques | |
| Méthotrexate | Méthotrexate, Ledertrexate |
| Pémétrexed | Alimta |
| Autres | |
| Hydroxyurée (hydroxycarbamide) | Hydréa |
ACTION DIRECTE SUR L’ADN OU LES ENZYMES ASSOCIÉES
Médicamentsalkylants(tableau8.3)
Ils remplacent un proton par un groupement alkyle. Extrêmement réactifs, ils peuvent produire des lésions covalentes, c’est-à-dire stables, entre les brins d’ADN, ce qui a pour effet d’entraver les processus de réplication et de transcription
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Moutardes à l’azote | |
| Busulphan Melphalan Chlorméthine Chlorambucil Cyclophosphamide Ifosfamide | Busilvex Alkéran Caryolysine Chloraminophène Endoxan Holoxan |
| Dérivés du platine | |
| Cisplatine Carboplatine Oxaliplatine | Cisplatine, Cisplatyl Carboplatine Eloxatine, Oxaliplatine |
| Nitrosourées | |
| Lomustine Carmustine Fotémustine Streptozotocine | Belustine Bicnu, Gliadel Muphoran Zanosar |
| Autres | |
| Mitomycine C Dacarbazine Estramustine Altrétamine Busulphan Procarbazine Témozolomide Thiotépa Pipobroman Trabectédine | Amétycine Déticène Estracyt Hexastat Myléran Natulan Témodal Thiotepa Vercyte Yondélis |
Médicaments intercalants
Les médicaments intercalants (tableau 8.4) se placent dans les sillons de l’ADN et bloquent sa réplication et sa transcription. Les deux chefs de file de cette famille sont l’adriamycine et la daunorubicine qui ont donné naissance au groupe des anthracyclines.
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Anthracyclines | |
| Doxorubicine Idarubicine Daunorubicine Épirubicine Pirarubicine | Adriblastine Zavedos Cérubidine, Daunoxome Farmorubicine Théprubicine |
| Anthracènediones | |
| Mitoxantrone | Novantrone |
Médicamentsscindants
lnhibiteursdestopo-isomérases(tableau 8.5)
Les topo-isomérases sont des enzymes clés dans les processus de réplication. Elles permettent de couper les brins d’ADN pour les dérouler (ADN-gyrases ou topo-isomérases II) et d’induire des coupures bicaténaires pour séparer les chromosomes avant la mitose (topo-isomérases I). Le produit le plus utilisé est l’irinotécan.
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Inhibiteurs de la topo-isomérase I | |
| Irinotécan Topotécan | Campto Hycamptin |
| Inhibiteurs de la topo-isomérase II (épipodophyllotoxines) | |
| Étoposide | Vépéside |
Poisonsdufuseaumitotique
Les poisons du fuseau mitotique (tableau 8.6) agissent de manière directe sur les molécules de tubuline indispensables à la constitution du fuseau mitotique et à la migration polaire des chromosomes pendant la mitose. On dispose de deux familles qui sont d’origine naturelle :
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Alcaloïdes de la pervenche | |
| Vinblastine Vindésine Vinorelbine Vincristine | Velbé Eldisine Navelbine, Vinorelbine Oncovin, Vincristine |
| Alcaloïdes de l’if (taxanes) | |
| Paclitaxel Docétaxel | Taxol Taxotère |
– les alcaloïdes de la pervenche (Vinca rosea) : vinblastine, vindésine, etc. ;
– les alcaloïdes de l’if (Taxus baccata) ou « taxanes » : paclitaxel, docétaxel.
Antiprotéines
La L-asparaginase (Kidrolase) est une enzyme injectable d’origine bactérienne, qui hydrolyse l’asparagine sanguine et provoque une carence en asparagine au niveau des cellules leucémiques.
Le bortézomib est un inhibiteur réversible et très sélectif du protéasome, impliqué dans la dégradation des protéines. Cette inhibition conduit à un arrêt du cycle cellulaire et à une mort cellulaire, encore appelée apoptose.
Modulateurs de la réponse biologique
IMMUNOMODULATEURS ET MODULATEURS HORMONAUX
De nombreuses classes de produits entrent dans cette catégorie. Elles sont regroupées dans les Tableau 8.7 and Tableau 8.10
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Modulateurs de la réponse de l’hôte | |
| BCG-thérapie lnterleukine-2 (IL-2) Interféron-a (lFN-α) | Immucyst Proleukin Roféron A |
| Antagonistes hormonaux et inhibiteurs de l’aromatase | |
| Dirigés « contre » les œstrogènes | |
| Antagonistes du récepteur des œstrogènes | |
| Fulvestrant Tamoxifène Torémifène | Faslodex Nolvadex Farestons |
| Inhibiteurs de l’aromatase (enzyme de synthèse des œstrogènes) | 0 |
| Anastrozole Exemestane Létrozole | Arimidex Aromastine Fémara |
| Dirigés « contre » la testostérone | |
| Antagoniste de la Gn-RH | |
| Dégarélix | Firmagon |
| Antagonistes de la testostérone | |
| Bicalutamide Cyprotérone Flutamide Nilutamide | Casodex Androcur Prostadirex Anandron |
| Agonistes hormonaux | |
| Agonistes de la LH-RH | |
| Buséréline Goséréline Leuproréline Triptoréline | Supréfact Zoladex Enantone Décapeptyl |
| Œstrogènes | |
| Diéthylstilbestrol | Distilbène |
| Progestatifs | |
| Médroxyprogestérone Mégéstrol | Dépo-Prodasone, Farlutal Mégace |
| Analogues de la somatostatine | |
| Lanréotide Octréotide | Somatuline Sandostatine |
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Inhibiteurs des tyrosine-kinases (cible entre parenthèses) | |
| Dasatinib (Bcr-Abl) Imatinib (Bcr-Abl) Nilotinib (Bcr-Abl) Erlotinib (HER1) Géfitinib (HER1) Lapatinib (HER1/HER2) Sorafénib (multikinases) Sunitinib (multikinases) | Sprycel Glivec Tasigna Tarcéva Iressa Tyverb Nexavar Sutent |
| Inhibiteur de la protéine-kinase mTOR | |
| Évérolimus Temsirolimus | Afinitor Torisel |
Les axes hormonaux sont particulièrement ciblés dans le cadre du traitement des cancers hormonodépendants de la prostate et du sein.
Les antiœstrogènes (tamoxifène) sont prescrits dans le cancer du sein lorsque le tissu tumoral est riche en récepteurs aux œstrogènes. Les dérivés de la progestérone sont utilisés dans le cancer du sein métastatique pour leurs effets antiprolifératifs.
Les antagonistes de la testostérone sont indiqués dans le cancer de la prostate simple ou métastasé.
THÉRAPEUTIQUES CIBLÉES
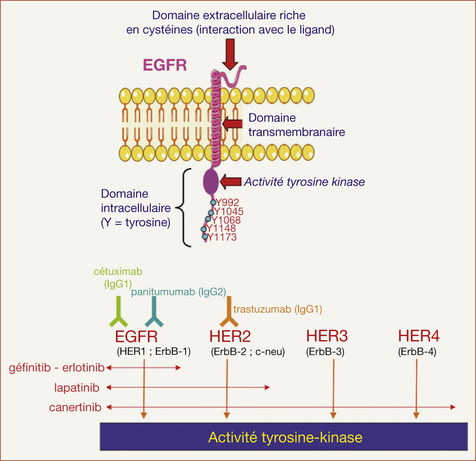
La dernière révolution dans le domaine des médicaments anticancéreux porte sur le ciblage spécifique de voies de transduction intracellulaires impliquées dans le développement de certains cancers. On y trouve des anticorps monoclonaux (tableau 8.8) et des i nhibiteurs de kinases (tableau 8.9). Une des dernières évolutions est celle des médicaments qui ciblent les récepteurs de l’EGF ou les kinases qui lui sont associées (figure 8.2).
| Dénomination commune internationale | Nom de spécialité |
|---|---|
| Anticorps monoclonaux dirigés contre les lymphocytes (cible entre parenthèses) | |
| Rituximab (CD20, lymphocytes B) Alemtuzumab (CD52, lymphocytes B et T) Catumaxomab (CD3, lymphocytes B) Ibritumomab (CD2, lymphocytes B) | Mabthera Mabcampath Removab Zévalin |
| Anticorps monoclonaux dirigés contre les récepteurs her (cible entre parenthèses) | |
| Cétuximab (EGFR, HER1) Panitumumab (EGFR, HER1) Trastuzumab (HER2) | Erbitux Vectibix Herceptin |
| Anticorps monoclonal dirigé contre le VEGF | |
| Bévacizumab (VEGF) | Avastin |
| HER = Human Estrogen Receptor, VEGF = Vascular Endothelial Growth Factor ou facteur de croissance vasculaire endothélial. | |
Effets indésirables et toxiques des chimiothérapies : aspects pratiques de leur gestion clinique
Toxicités aiguës
L’incidence et la sévérité dépendent de la dose administrée et des modalités d’administration. Certaines peuvent être prévenues par des mesures appropriées (voir tableau 8.11). Les toxicités aiguës sont en général réversibles, elles apparaissent quelques heures à quelques jours après le début du traitement.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


