C. Coulon, Ph. Bourgeot, Y. Robert, P. Vaast, V. Debarge and B. Guérin La région craniofaciale est une région complexe, faisant intervenir les voies aériennes et digestives, le système nerveux central et les organes des sens. Des tissus d’origine embryologique différente, ectodermique, endodermique et mésodermique, ainsi que la population craniofaciale des cellules des crêtes neurales y participent. Cette complexité explique la fréquence élevée des malformations craniofaciales. Cette région se forme entre les 3e et 8e semaines du développement. Elle nécessite une synergie temporo-spatiale des phénomènes cellulaires, la croissance adéquate et simultanée des cinq bourgeons de la face et du cerveau et des interactions de l’endoblaste et de l’ectoblaste avec la matrice extracellulaire. Elle implique le rôle d’induction du système nerveux central, les propriétés du liquide amniotique, qui joue un rôle tensio-actif et intervient sur le modelage externe, ainsi que l’activité succion–déglutition à partir du 3e mois. La formation du crâne comprend : Pour comprendre ce type de malformations, il faut rappeler que la lèvre supérieure, les quatre incisives supérieures et le palais primaire dérivent de la même structure embryologique (le bourgeon frontal), alors que les palais osseux et vélaire sont issus des bourgeons maxillaires. Si la lèvre est fermée, le palais primaire et l’arc alvéolaire médian le seront donc également ; si la lame palatine est vue intacte, le palais osseux secondaire est bien fermé ; si la luette est visible, l’ensemble du palais secondaire est intact. Des causes infectieuses sont décrites (rubéole) ainsi que des agents tératogènes (acide rétinoïde, acide valproïque, hydantoïne, alcool). On distingue les fentes labiales ou labionarinaires (25 %), les fentes labiopalatines (50 %) et les fentes palatines (25 %). Ces dernières sont de diagnostic prénatal extrêmement difficile surtout si elles sont isolées. Elles sont le plus souvent unilatérales (75 %), parfois bilatérales et parfois décrites « médianes » (correspondant à une agénésie du processus prémaxillaire). Les fentes bilatérales et « médianes »doivent rendre particulièrement attentive l’étude cérébrale car elles peuvent s’intégrer dans une pathologie de la ligne médiane (holoprosencéphalie notamment, voir chap. 8). Les anomalies chromosomiques associées sont extrêmement rares en cas de fentes labiales ou labiopalatines isolées, mais le caractère isolé en anténatal étant difficile à affirmer conduit de nombreuses équipes à proposer un contrôle du caryotype fœtal en cas de fente diagnostiquée. Le diagnostic échographique initial est porté sur une interruption de la lèvre supérieure sur la coupe « nez–bouche » recommandée par le CNTE (voir fig. 6.12). L’analyse échographique d’une fente doit comprendre : Le diagnostic échographique en est généralement simple lors de l’échographie de 22 SA, en l’absence d’interposition des mains ou du cordon ombilical, ou d’une tête fixée dans une mauvaise orientation. En échographie 2D, les trois plans de coupe sont utiles pour une analyse complète : Une étude en 3D, quand elle est réalisable, donnera une idée plus précise et réaliste de la dysmorphie. Le mode surfacique permet l’étude de la fente labionarinaire, le mode triplan permettra une reconstruction de l’interface du palais secondaire lorsque l’étude 2D est impossible (fig. 9.3i et j). Cependant, il est nécessaire d’informer les parents de cette étude 3D-4D qui peut parfois choquer. L’échographiste peut également acquérir les volumes, les retravailler secondairement et les présenter aux parents lors d’une consultation suivante. Ces reconstructions en 3D peuvent être utiles au chirurgien plasticien pour guider son entretien prénatal (fig. 9.4). En cas de fente bilatérale (fig. 9.5b et c), le bourgeon médian peut réaliser une masse centrale saillante visible sur une coupe sagittale (fig. 9.5a) ou transversale (fig. 9.6). Le profil est nettement perturbé par le bourgeon prémaxillaire qui fait saillie vers l’avant et surplombe la gencive supérieure en retrait. Cet aspect ne doit pas être confondu avec un tératome nasopharyngé extériorisé ou un proboscis (voir plus loin). Les reconstructions 3D, quand c’est possible, viendront compléter le bilan anatomique (fig. 9.7). Une fente médiane est beaucoup plus rare (fig. 9.8). Elle traduit une anomalie de développement du bourgeon fronto-nasal normalement induit par le cerveau et doit faire évoquer une anomalie cérébrale au niveau de la ligne médiane. Elle peut donc être associée à : Tous ces éléments doivent faire évoquer une anomalie chromosomique, surtout la trisomie 13. L’amniocentèse est proposée aux parents pour étude du caryotype fœtal et recherche du syndrome de DiGeorge en cas de fente palatine associée, mais elle peut se discuter en cas d’antécédent familial du 1er degré (car le risque chromosomique devient extrêmement faible). En revanche, celui-ci est beaucoup plus important en cas de fente associée (surtout les fentes bilatérales et médianes) à type de trisomie 13 et trisomie 18. La difficulté de pouvoir affirmer en anténatal le caractère isolé d’une fente conduit la majorité des équipes à proposer cet examen systématiquement. En effet, 12 à 22 % des fentes dites isolées en anténatal se révèlent associées à d’autres anomalies en post-natal. Au cours de la grossesse, la tolérance de cette malformation est bonne mais un hydramnios ou un excès de liquide amniotique, par trouble de la déglutition, est habituellement retrouvé dans les larges fentes labiopalatines. L’analyse du profil fœtal nécessite une coupe sagittale médiane stricte qui n’est pas toujours facile à obtenir : attention aux fausses impressions ! En cas de doute, il est possible de mesurer l’angle facial supérieur (entre la ligne droite tangente au front et la ligne droite tangente aux OPN, valeur normale 131° ± 13°). L’angle facial inférieur peut également être calculé (entre la ligne droite perpendiculaire au front et la ligne droite formée par le menton et la lèvre la plus protruse, valeur normale 64 à 68° ± 8°) (voir fig.6.6c). Des OPN absents ou nettement inférieurs à 2,5 mm à partir de 20 SA sont suspects (fig. 9.9, et voir chap. 7 et voir chap. 16, fig. 16.30). Il s’agit d’un signe d’appel majeur de trisomie 21 (62 % des enfants T21 ont une hypoplasie des OPN contre 1,2 % des fœtus normaux) permettant de proposer un contrôle du caryotype fœtal même si ce signe est isolé. Il convient néanmoins de veiller à ce que le faisceau ultrasonore soit le plus perpendiculaire possible à l’axe formé par les OPN pour obtenir une image satisfaisante. Par exemple, la mesure sera impossible sur un fœtus dont la tête est très défléchie. L’hypoplasie peut aussi être un signe d’appel d’une dysplasie squelettique rare telle que la dysostose cléidocrânienne, où il existe une aplasie ou hypoplasie claviculaire, ou également la dysplasie thanatophore. La fœtopathie warfarinique (warfarin syndrome) peut également être responsable d’une hypoplasie des OPN (associée à un RCIU, un aplatissement de l’ensellure nasale, des ponctuations épiphysaires – voir fig. 15.23). Ce signe est habituellement décrit dans l’achondroplasie (voir fig.15.18 et 15.19), le nanisme thanatophore (voir fig.15.8) et le syndrome d’Apert. L’appréciation est assez subjective et il existe des variations interindividuelles à ce niveau. Cependant, les formes pathologiques sont souvent caricaturales. Elle s’observe dans le cadre d’une holoprosencéphalie semi- ou plus volontiers alobaire et se traduit : Il se caractérise par le recul du menton en arrière du plan tangent à la partie inférieure de l’os frontal. Sur une coupe frontale de la face, on ne parvient pas à visualiser le menton dans le même plan que le nez et la bouche. La mesure de l’angle facial inférieur peut permettre d’objectiver cette impression visuelle. Le rétrognathisme est caractéristique de la séquence (ou syndrome) de Pierre Robin mais on le rencontre aussi dans de nombreux syndromes ou associations malformatives (fig. 9.12 et 9.13a et b) tels que le syndrome de Cornelia de Lange (fig. 9.13c à e), la trisomie 18 et la dysostose craniofaciale.
Pathologie cervicofaciale
 la pathologie faciale est parfois le signe d’appel d’une malformation plus complexe ou peut s’intégrer dans une malformation syndromique dont le pronostic pourrait s’en trouver alourdi ;
la pathologie faciale est parfois le signe d’appel d’une malformation plus complexe ou peut s’intégrer dans une malformation syndromique dont le pronostic pourrait s’en trouver alourdi ;
Rappel embryologique11
 la base du crâne ou chondrocrâne, issu du mésenchyme préchordal qui donne les os de la base du crâne et les capsules sensorielles,
la base du crâne ou chondrocrâne, issu du mésenchyme préchordal qui donne les os de la base du crâne et les capsules sensorielles,
Pathologie de la face
Fentes labiopalatines
 son caractère uni- ou bilatéral ;
son caractère uni- ou bilatéral ;
 la mesure de la largeur de la fente ;
la mesure de la largeur de la fente ;
 la déviation de la pointe du nez ;
la déviation de la pointe du nez ;
 l’étude du profil et de la déviation plus ou moins prononcée du bourgeon latéral ;
l’étude du profil et de la déviation plus ou moins prononcée du bourgeon latéral ;
Diagnostic échographique
Fente labiale ou labiopalatine unilatérale
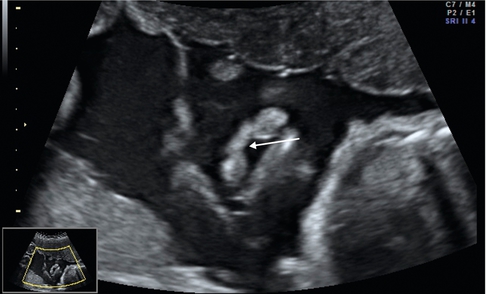
 la coupe frontale (fig. 9.1) permet de mettre en évidence une discontinuité de la lèvre supérieure et servira d’image de référence. Il est possible sur ce plan de coupe de mesurer la largeur de la fente labiale et d’étudier la déviation de la pointe du nez (notions importantes à transmettre au chirurgien plasticien pour l’entretien prénatal). L’ouverture de la gencive et de l’arcade dentaire peut aussi être visible sur la coupe frontale ;
la coupe frontale (fig. 9.1) permet de mettre en évidence une discontinuité de la lèvre supérieure et servira d’image de référence. Il est possible sur ce plan de coupe de mesurer la largeur de la fente labiale et d’étudier la déviation de la pointe du nez (notions importantes à transmettre au chirurgien plasticien pour l’entretien prénatal). L’ouverture de la gencive et de l’arcade dentaire peut aussi être visible sur la coupe frontale ;
a. 24 SA. Fente unilatérale, labionarinaire, fine (→).
b. 34 SA. Fente labionarinaire unilatérale (→).
c. 24 SA. Mesure (1) de la largeur d’une fente unilatérale.
d. Fente labiale à 22 SA avec rétraction de la lèvre et déviation de la pyramide nasale.
e. Fente labiomaxillaire : le plan frontal montre à la fois la fente labiale (d’une largeur de 8 mm) et la fente maxillaire (→) au niveau de l’arcade dentaire.
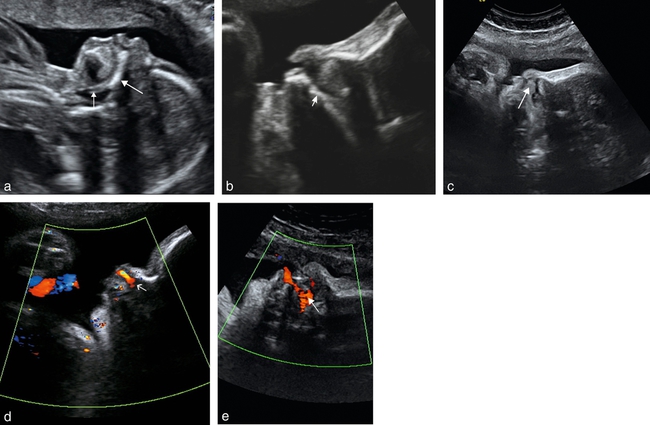
 la coupe sagittale (fig. 9.2) permet d’observer une déformation lorsque le bourgeon médian est projeté en avant de façon importante. Elle permet, également, de montrer des signes indirects de fente palatine associée, étude à réaliser de façon dynamique : ascension de la langue dans les fosses nasales lors des mouvements de déglutition, mélange des flux buccal et nasal au Doppler couleur, même si ce signe peut être absent en cas de petite fente palatine associée, obstruée par la langue ascensionnée ;
la coupe sagittale (fig. 9.2) permet d’observer une déformation lorsque le bourgeon médian est projeté en avant de façon importante. Elle permet, également, de montrer des signes indirects de fente palatine associée, étude à réaliser de façon dynamique : ascension de la langue dans les fosses nasales lors des mouvements de déglutition, mélange des flux buccal et nasal au Doppler couleur, même si ce signe peut être absent en cas de petite fente palatine associée, obstruée par la langue ascensionnée ;
a. 18 SA. Coupe sagittale permettant la visualisation d’un palais osseux normal sous la forme d’une ligne continue large et échogène (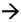 ), prolongée par le palais vélaire (→).
), prolongée par le palais vélaire (→).
b. Sous le nez, on retrouve un « décrochage » qui attire l’attention (fœtus de 5 mois). Le palais osseux (→) semble intact.
c. 33 SA. Signe indirect de fente palatine avec ascension de la langue (→) dans les fosses nasales.
d. 26 SA. En mode Doppler couleur, une expiration vigoureuse donne un flux nasal net (→) sans aucun flux dans la bouche : absence probable de fente palatine.
e. 33 SA. Signe indirect de fente palatine associée avec mélange des flux buccal et nasal (→).
 la coupe transversale (fig. 9.3a à d) permet de rechercher une atteinte gingivo-alvéolaire associée et également des signes indirects de fente palatine lorsque la langue, échogène, est retrouvée au niveau du defect maxillaire (fig. 9.3e) et que la visualisation directe de la lame palatine postérieure est impossible (fig. 9.3f). Lorsqu’elle est retrouvée, ceci traduit l’intégrité du palais osseux secondaire. Cette image rectiligne hyperéchogène correspond à l’interface palais osseux/palais vélaire. Il est également possible de visualiser les apophyses ptérygoïdes de part et d’autre de cette image (voir fig. 6.10a). L’étude du palais se fera sur un fœtus face en avant, au mieux tête défléchie (ne pas hésiter à le mobiliser pour lui amener le visage contre la sonde d’échographie). Certains auteurs ont récemment montré la possibilité de rechercher l’atteinte du voile du palais (fig. 9.3g) et la présence de luette bifide, mais cette étude reste aujourd’hui relativement difficile. La coupe transversale permet également l’étude de la déviation de la pointe du nez (fig. 9.3h).
la coupe transversale (fig. 9.3a à d) permet de rechercher une atteinte gingivo-alvéolaire associée et également des signes indirects de fente palatine lorsque la langue, échogène, est retrouvée au niveau du defect maxillaire (fig. 9.3e) et que la visualisation directe de la lame palatine postérieure est impossible (fig. 9.3f). Lorsqu’elle est retrouvée, ceci traduit l’intégrité du palais osseux secondaire. Cette image rectiligne hyperéchogène correspond à l’interface palais osseux/palais vélaire. Il est également possible de visualiser les apophyses ptérygoïdes de part et d’autre de cette image (voir fig. 6.10a). L’étude du palais se fera sur un fœtus face en avant, au mieux tête défléchie (ne pas hésiter à le mobiliser pour lui amener le visage contre la sonde d’échographie). Certains auteurs ont récemment montré la possibilité de rechercher l’atteinte du voile du palais (fig. 9.3g) et la présence de luette bifide, mais cette étude reste aujourd’hui relativement difficile. La coupe transversale permet également l’étude de la déviation de la pointe du nez (fig. 9.3h).
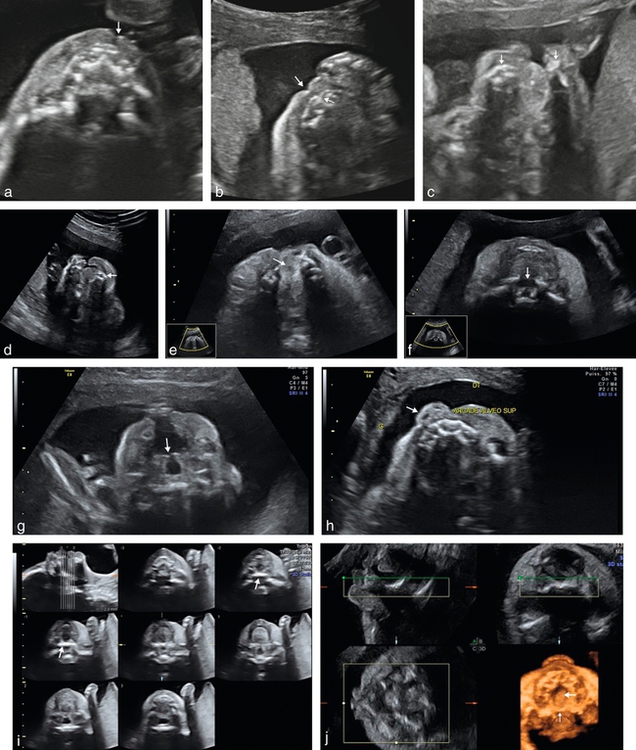
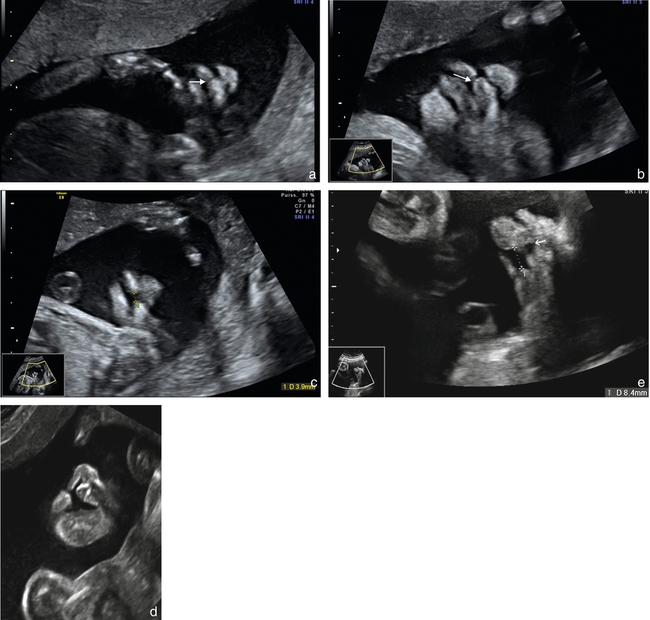
a. 23 SA. Petite fente labiale (→) avec respect de l’arc alvéolaire supérieur.
b. 25 SA. Visualisation d’une petite fente labiale ( ) avec une discrète encoche sur l’arc alvéolaire supérieur (→) (encoche gingivale ?).
) avec une discrète encoche sur l’arc alvéolaire supérieur (→) (encoche gingivale ?).
c. 23 SA. Large fente labiopalatine, ascension de la langue (*) au travers de la fente palatine, remarquer le retrait latéral des deux parties alvéolaires (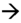 ) qui ne se trouvent pas en regard de l’ouverture labiale (ouverture alvéolaire plus large que l’ouverture labiale).
) qui ne se trouvent pas en regard de l’ouverture labiale (ouverture alvéolaire plus large que l’ouverture labiale).
d. 24 SA. Large fente labiopalatine, retrait latéral et postérieur de la partie latérale de l’arc alvéolaire ( ).
).
e. 33 SA. Signe indirect de fente palatine associée à une fente labionarinaire, avec ascension de la langue au travers de la fente palatine (→).
f. 25 SA. Fente palatine (fente labiopalatine bilatérale) évoquée par la rupture de la lame palatine postérieure (→).
g. Même foetus que f fente labiopalatine bilatérale, avec division du palais vélaire visible sur cette incidence (→).
h. 25 SA. Fente labionarinaire unilatérale, déviation discrète de la pointe du nez (→).
i. 3D triplan, mode TUI, reconstruction de la lame palatine postérieure (→). Acquisition sur un fœtus de profil strict, tête défléchie.
j. Palais ogival. Reconstruction 3D triplan, intégrité de la lame palatine postérieure (→), mais aspect creusé du palais ( ).
).
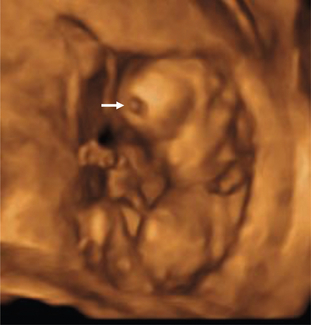
a. 24 SA. Fente labiale unilatérale (encoche de la lèvre supérieure →), reconstruction 3D surface.
b. 24 SA. Large fente labiopalatine unilatérale (→), reconstruction 3D surface.
c. 34 SA. Fente labionarinaire unilatérale (→), déviation discrète de la pointe du nez, reconstruction 3D surface.
d. Fente labiopalatine unilatérale avec rétraction vers la gauche de la lèvre et du nez.
e. 24 SA. Fente labionarinaire unilatérale (→). Reconstruction 3D surface.
f. 29 SA. Fente labiopalatine unilatérale, fœtus bouche ouverte, permettant de visualiser la fente palatine associée (→), reconstruction 3D surface.
g. Nouveau-né à terme. Fente labiopalatine avec l’ouverture du palais bien visible.
Fente labiale bilatérale
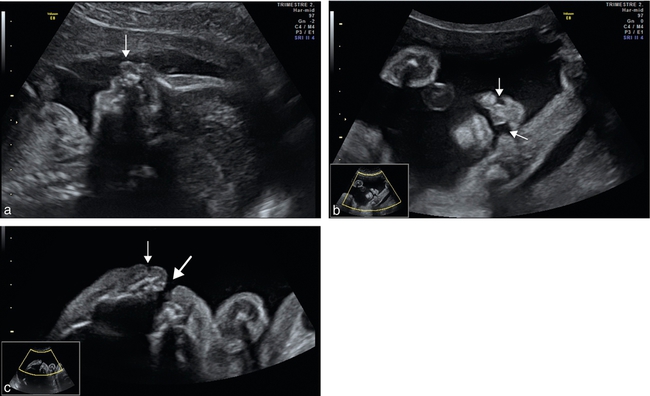
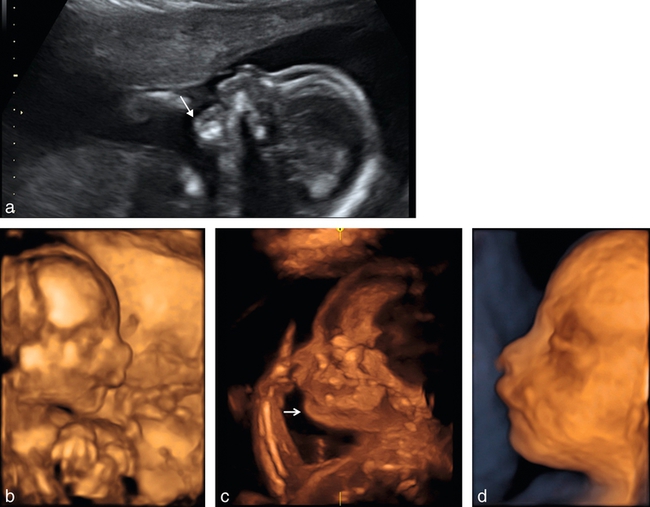
a. 24 SA. Fente bilatérale, vue de profil, visualisation du bourgeon médian qui fait saillie sous le nez (→).
b. 25 SA. Fente bilatérale (→), coupe frontale, mode bidimensionnel. Noter la fente plus large d’un côté (à droite de l’image) et plus fine de l’autre. Peu ou pas de déviation de la pointe du nez.
c. 25 SA. Fente bilatérale, coupe transversale. Le côté droit (→) montre une fente labiale avec l’encoche labiale, le côté gauche ( ) montre une fente labiopalatine large.
) montre une fente labiopalatine large.
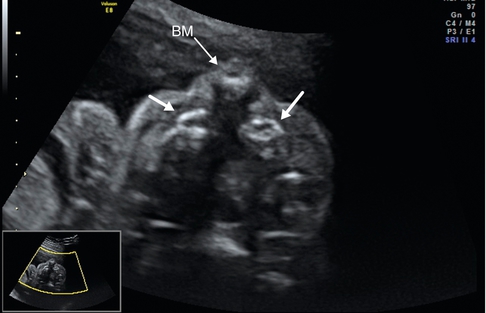
26 SA. Coupe transversale, fente labiopalatine bilatérale, visualisation du bourgeon médian projeté vers l’avant (BM) et des parties latérales de l’arcade maxillaire supérieure décalées vers l’arrière (→).
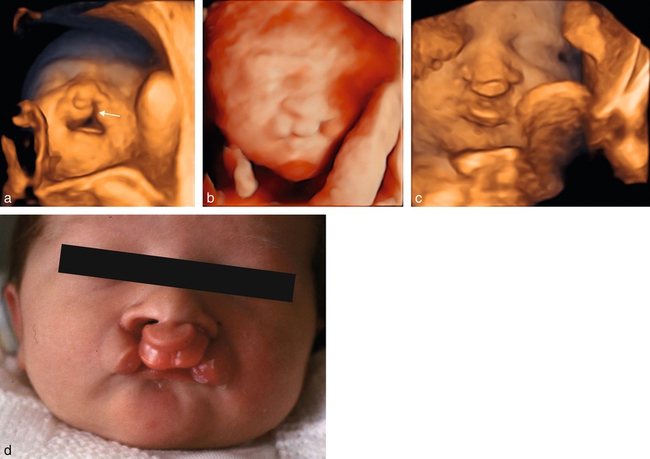
a. 25 SA. Fente labiopalatine bilatérale asymétrique. Mode 3D surfacique, fœtus tête en extension, bouche ouverte. Cette position permet une visualisation directe de la fente palatine (→).
b. Même patiente, mode 3D surfacique, rendu HD-live.
c. 33 SA. Fente bilatérale, mode 3D surfacique.
d. Fente labiale avec bourgeon labiomaxillaire médian.
Fente médiane
 une malformation nasale : nez petit, une seule narine, voire un proboscis ;
une malformation nasale : nez petit, une seule narine, voire un proboscis ;
 des anomalies oculaires : microphtalmie, cyclopie ;
des anomalies oculaires : microphtalmie, cyclopie ;
Prise en charge
Anomalies du profil fœtal et du nez
Hypoplasie des os propres du nez (OPN)
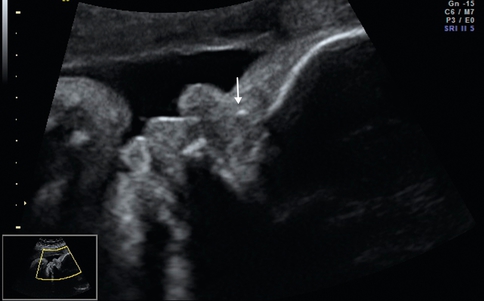
Coupe sagittale du profil, os propres du nez punctiformes (→). Trisomie 21.
Ensellure nasale marquée
Absence de nez, arhinie ou arhinencéphalie
 soit par l’absence totale de nez (fig. 9.10a à d) avec souvent présence d’un bourgeon au-dessus du plan orbitaire correspondant au proboscis (fig. 9.10e et fig. 9.11) en cas de cyclopie (orbite unique) et d’ethmocéphalie (hypotélorisme majeur) ;
soit par l’absence totale de nez (fig. 9.10a à d) avec souvent présence d’un bourgeon au-dessus du plan orbitaire correspondant au proboscis (fig. 9.10e et fig. 9.11) en cas de cyclopie (orbite unique) et d’ethmocéphalie (hypotélorisme majeur) ;
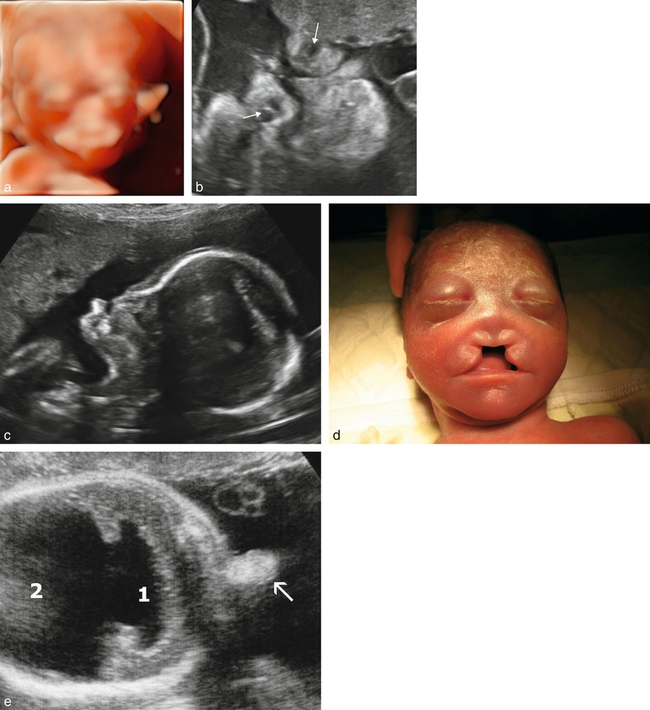
a. 12 SA. Arhinie, fente « médiane » avec agénésie du prémaxillaire et arhinie, hypotélorisme. Fœtus présentant une holoprosencéphalie alobaire. Mode 3D surfacique, rendu HD-live.
b. 26 SA. Arhinie, hypotélorisme et exophtamie (flèches marquant les yeux). Holoprosencéphalie.
c. Arhinencéphalie (23 SA) associée à une holoprosencéphalie semi-lobaire, coupe du profil. Disparition du massif médian avec absence de la lèvre supérieure et de la pyramide nasale.
d. Arhinencéphalie (23 SA). « Avortement » du massif médian de la face.
e. Proboscis (22 SA), coupe transversale. Appendice médian sus-orbitaire (→). Holoprosencéphalie alobaire avec cavité ventriculaire unique (1) et volumineux sac dorsal (2).
 soit par l’existence d’une pyramide nasale rudimentaire avec une seule narine (cébocéphalie).
soit par l’existence d’une pyramide nasale rudimentaire avec une seule narine (cébocéphalie).
Rétrognathisme
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


9: Pathologie cervicofaciale