Chapitre 8
Anomalies cranio-encéphaliques
Ph. Bourgeot, J. Bigot, S. Joriot and D. Parzy
Anomalies de la biométrie céphalique
Le dépistage de ces anomalies nécessite une mesure correcte du diamètre bipariétal (DBP ou BIP) et de la circonférence céphalique (CC ou PC).
La méthode de mesure utilisée est celle recommandée par le Collège français d’échographie fœtale (voir chap. 7, Biométrie fœtale). Le DBP se mesure sur une coupe transversale de la tête fœtale (coupe transthalamique) occupant plus de la moitié de la taille totale de l’image où doivent être vus la scissure interhémisphérique, le cavum du septum pellucidum, les thalamus, le troisième ventricule et la citerne ambiante (citerne du mésencéphale). La symétrie doit être respectée de part et d’autre de la ligne médiane. Théoriquement les cornes occipitales ne sont pas vues sur cette coupe (en pratique la mesure est acceptable). En revanche, la fosse cérébrale postérieure avec le cervelet ne doit pas être visualisée. Le caliper d’entrée est placé au milieu de l’os pariétal proximal, le caliper de sortie au niveau de l’os pariétal distal. L’axe de la mesure est perpendiculaire à la scissure interhémisphérique. La CC se mesure dans le plan du DBP (en conservant les mêmes critères de qualité) par une ellipse dont les points sont placés à la périphérie du pourtour osseux.
Les mesures sont exprimées en millimètres et réparties en percentiles (3–10–20–50–90–97) pour chaque semaine d’aménorrhée (voir tableau 7.1 et 7.2).
Microcéphalie
Sa fréquence est estimée de 1 à 4 pour 10 000 naissances. Quatorze pour cent des microcéphalies sont diagnostiquées en anténatal. Ce sont les microcéphalies primitives par développement anormal du cerveau durant les sept premiers mois de grossesse (anomalie de la migration et de la prolifération neuronale). Les microcéphalies secondaires à une « agression » cérébrale survenant en fin de grossesse (infectieuse, hémorragique, hypoxique…) ne sont diagnostiquées qu’en période péri- ou néonatale. Anatomiquement, on parle de microcéphalie lorsque le périmètre crânien est à moins 3 déviations standard (DS) au-dessous de la moyenne. Pour envisager en période anténatale ce diagnostic lourd de conséquence, il faut que le DBP et le PC soient nettement inférieurs au 3e percentile de l’âge gestationnel considéré. Si le DBP est anormal mais que le PC reste dans des valeurs acceptables il s’agit d’une dolichocéphalie simple par modelage de la tête fœtale (présentation du siège, oligoamnios…) qui isolément n’a pas de caractère pathologique.
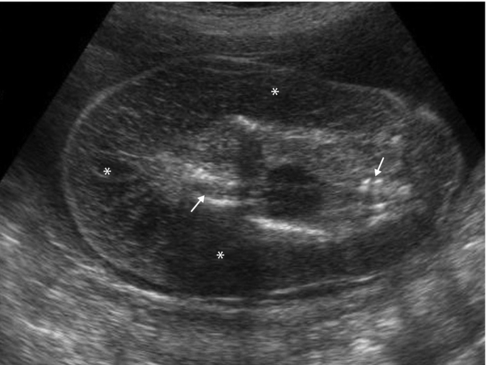
Le diagnostic est rarement posé avant 22 SA mais il peut être facile, si les mesures abdominales et osseuses sont normales et si l’âge de la grossesse est connu avec précision. Le pôle céphalique peut présenter une forme inhabituelle et sur une coupe de profil le front est parfois fuyant lié à une hypotrophie des lobes frontaux. Il reste néanmoins à éliminer une craniosynostose à type de turicéphalie (voir plus loin). Les espaces péricérébraux peuvent être larges. L’existence d’une ventriculomégalie associée ou d’anomalies de giration viendront conforter le diagnostic (fig. 8.1). L’établissement d’une courbe de croissance permet de noter parfois un infléchissement de celle-ci qui reste souvent progressif, modéré et tardif. L’IRM en évaluant plus précisément les mesures exactes du cerveau confirme la suspicion diagnostique.
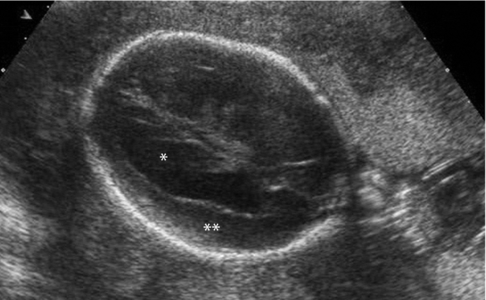
Le PC est mesuré à 202 mm, ce qui est nettement inférieur au 3e percentile qui est de 215 mm. Il existe en plus une ventriculomégalie (∗) et la vallée sylvienne n’est pas individualisée (∗ ∗).
Le diagnostic est en revanche difficile si toutes les mesures fœtales sont faibles mais sans malformation échographiquement décelable. On évoquera alors plus volontiers une erreur de datation, un retard de croissance intra-utérin symétrique (harmonieux) mais aussi un fœtus constitutionnellement petit. Dans cette dernière hypothèse, les mesures sont faibles mais la croissance reste partiellement conservée. Il faut penser à vérifier systématiquement le morphotype parental (notamment le PC) et les antécédents (poids, taille, PC et sexe des enfants précédents).
 malformations diverses : holoprosencéphalie, céphalocèle, troubles de la migration avec ventriculomégalie, dysgénésie du corps calleux et lissencéphalie le plus souvent dans le cadre d’un syndrome polymalformatif (lissencéphalie de type I et III, syndrome de Smith-Lemli-Opitz, syndrome de Dubowitz et ostéochondrodysplasies) ;
malformations diverses : holoprosencéphalie, céphalocèle, troubles de la migration avec ventriculomégalie, dysgénésie du corps calleux et lissencéphalie le plus souvent dans le cadre d’un syndrome polymalformatif (lissencéphalie de type I et III, syndrome de Smith-Lemli-Opitz, syndrome de Dubowitz et ostéochondrodysplasies) ;
 aberrations chromosomiques fréquentes : trisomie 9, 13, 22 et diverses délétions (syndrome de Wolf-Hirschhorn) ;
aberrations chromosomiques fréquentes : trisomie 9, 13, 22 et diverses délétions (syndrome de Wolf-Hirschhorn) ;
 microcéphalie à giration simplifiée par un arrêt précoce de la migration et de la prolifération neuronales s’associant à une lissencéphalie et une hypoplasie du corps calleux, alors que dans la microcéphalie vera il n’existe pas d’anomalie anatomique cérébrale notable et la giration est à peu près normale ;
microcéphalie à giration simplifiée par un arrêt précoce de la migration et de la prolifération neuronales s’associant à une lissencéphalie et une hypoplasie du corps calleux, alors que dans la microcéphalie vera il n’existe pas d’anomalie anatomique cérébrale notable et la giration est à peu près normale ;
 causes infectieuses ou métaboliques : CMV, rubéole, herpès, syndrome d’alcoolisation fœtale, cocaïne, phénylcétonurie…
causes infectieuses ou métaboliques : CMV, rubéole, herpès, syndrome d’alcoolisation fœtale, cocaïne, phénylcétonurie…
En cas de lésions clastiques précoces et importantes (aprosencéphalie, atélencéphalie), on peut observer un chevauchement des sutures.
Macrocéphalie
On parle de macrocéphalie lorsque le périmètre céphalique est à plus 3 DS au-dessus de la moyenne. On peut distinguer la macrocrânie où le PC osseux est à plus 3 DS et la mégalencéphalie qui se caractérise anatomiquement par un poids excessif du cerveau de plus 2 DS.
En échographie, le DBP et le PC sont nettement supérieurs au 97e percentile pour l’âge gestationnel en discordance avec le reste de la biométrie, ce qui exclut une erreur de datation. Si toutes les mesures fœtales sont nettement supérieures aux normes, on peut discuter une erreur de datation, une macrosomie constitutionnelle ou d’origine diabétique.
 anomalie squelettique (ostéochondrodysplasie, dysostose) ;
anomalie squelettique (ostéochondrodysplasie, dysostose) ;
 hydrocéphalie de différentes origines ;
hydrocéphalie de différentes origines ;
 hydranencéphalie par destruction ischémique ;
hydranencéphalie par destruction ischémique ;
 troubles de la migration neuronale :
troubles de la migration neuronale :
 syndrome de Walker-Warburg : lissencéphalie de type II, hydrocéphalie, hétérotopie cérébelleuse, ± syndrome de Dandy-Walker, céphalocèle (et décollement rétinien),
syndrome de Walker-Warburg : lissencéphalie de type II, hydrocéphalie, hétérotopie cérébelleuse, ± syndrome de Dandy-Walker, céphalocèle (et décollement rétinien),
 hémimégalencéphalie avec ventricules plus ou moins dilatés et déformés : syndromes neurocutanés (Protée, Klippel-Trenaunay, neurofibromatose de type I…),
hémimégalencéphalie avec ventricules plus ou moins dilatés et déformés : syndromes neurocutanés (Protée, Klippel-Trenaunay, neurofibromatose de type I…),
 mégalencéphalie symptomatique : dilatation ventriculaire, corps calleux quelquefois épaissi, dysplasie corticale et operculation incomplète,
mégalencéphalie symptomatique : dilatation ventriculaire, corps calleux quelquefois épaissi, dysplasie corticale et operculation incomplète,
 overgrowth syndromes (Sotos, Weaver),
overgrowth syndromes (Sotos, Weaver),

L’IRM permet de différencier cerveau et espaces péricérébraux, de vérifier la giration (pachymicrogyrie), de rechercher des hétérotopies de la substance blanche ou une dysplasie corticale.
Anomalies des contours du crâne
Acrânie
 l’absence totale de voûte et d’encéphale, avec une face rudimentaire (anencéphalie) (fig. 8.2) ;
l’absence totale de voûte et d’encéphale, avec une face rudimentaire (anencéphalie) (fig. 8.2) ;
 à la présence d’un encéphale anormal bombant dans le liquide amniotique (exencéphalie) et se nécrosant par la suite (au contact avec le liquide amniotique et/ou par frottements de l’encéphale contre la paroi) ;
à la présence d’un encéphale anormal bombant dans le liquide amniotique (exencéphalie) et se nécrosant par la suite (au contact avec le liquide amniotique et/ou par frottements de l’encéphale contre la paroi) ;
 jusqu’à l’acalvaria plus ou moins totale par défaut de migration mésenchymateuse. Le cerveau, normal n’est recouvert que par de la peau, l’os et la dure-mère étant absents.
jusqu’à l’acalvaria plus ou moins totale par défaut de migration mésenchymateuse. Le cerveau, normal n’est recouvert que par de la peau, l’os et la dure-mère étant absents.
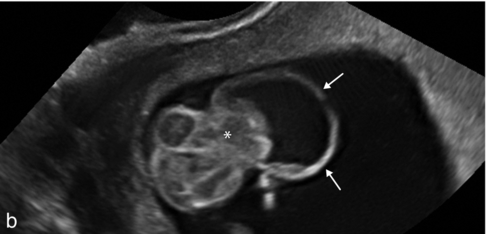
En échographie, le diagnostic est possible dès 12–13 SA et se caractérise par un pôle céphalique anormal (structure de l’encéphale inhabituelle, désorganisée et absence de ligne interhémisphérique) recouvert parfois par une membrane méningée (fig. 8.3). Il s’agit au départ plutôt d’un aspect d’exencéphalie (fig. 8.4). Le tissu cérébral anormal, bombant dans le liquide amniotique, va se lyser et donner au 2e trimestre une anencéphalie (fig. 8.5). On note alors l’absence de voûte crânienne visible et un DBP impossible à mesurer. La base du crâne et la face sont identifiables mais les orbites paraissent volumineuses. Le liquide amniotique est finement échogène.
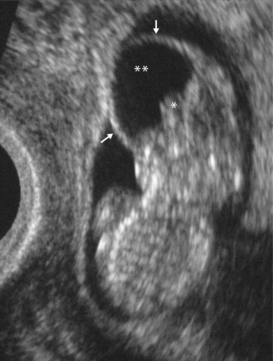
L’encéphale (∗), anormal et le liquide cérébrospinal (∗∗) sont recouverts par une membrane méningée épaisse (→).
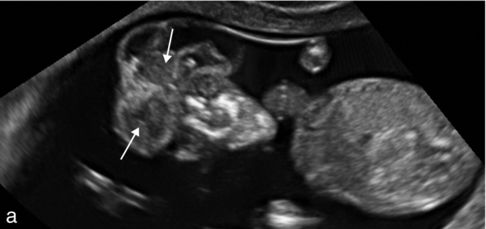
a. Désorganisation des structures cérébrales sus-orbitaires (→) qui ne sont pas recouvertes par la voûte crânienne, absente. L’évolution peut se faire vers une véritable anencéphalie suite à la destruction des structures cérébrales au contact du liquide amniotique.
b. La coupe transversale passe au niveau de l’exencéphalie (∗). On distingue des éléments échogènes (encéphaliques), anéchogènes (liquide cérébrospinal) ; l’ensemble est partiellement entouré des méninges (→).
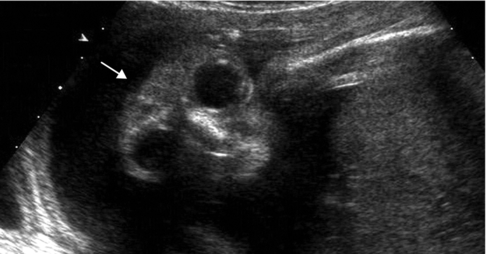
Coupe frontale de la face. Les orbites sont marquées et il n’existe ni encéphale ni voûte crânienne au-dessus des orbites (→).
Des malformations associées peuvent être retrouvées : rachischisis (25 % des cas), fente labiopalatine (10 %), malformations urinaires (16 %), digestives (6 %), cardiaques (4 %). L’hydramnios est fréquent par troubles de déglutition. Les mouvements fœtaux paraissent saccadés et amples provoqués par le contact du moignon céphalique avec l’utérus.
Dans l’acalvaria (c’est-à-dire l’absence de voûte crânienne) qu’elle soit totale (fig. 8.6) ou partielle, l’encéphale extériorisé garde un aspect homogène et symétrique (car il est protégé par un tissu méningé et cutané) tout à fait différent de l’exencéphalie où il est destructuré et plus ou moins détruit.
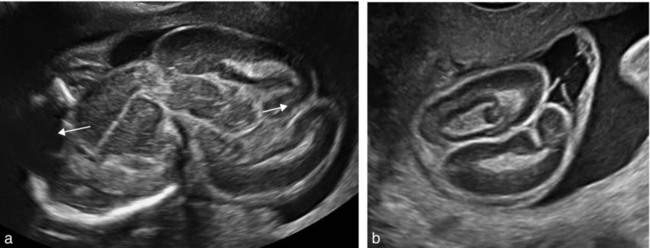
a. 22 SA. Coupe frontale du pôle céphalique. Il n’y a pas de voûte crânienne individualisable. Les hémisphères cérébraux gardent une certaine symétrie mais sont étalés latéralement (→).
b. Même fœtus : l’absence de désorganisation des structures cérébrales est encore plus nette sur cette coupe transversale du pôle céphalique. Source : Dr C. Coulon.
Certaines curiosités embryologiques et échographiques ou syndromes rares peuvent se rencontrer :
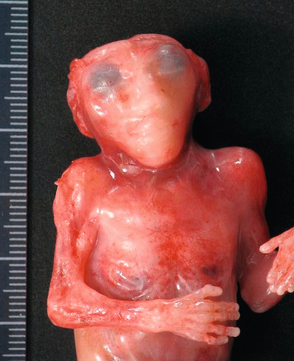
 l’exencéphalie par maladie des brides amniotiques. L’encéphale extériorisé et anormal est adhérent au placenta, fixé lors des mouvements fœtaux (fig. 8.7). Dans cette forme, on retrouve une dysmorphie faciale complexe à type de fentes faciales asymétriques et obliques, d’hypertélorisme, d’anomalie de l’ensellure nasale, de micro- ou anophtalmie unilatérale ;
l’exencéphalie par maladie des brides amniotiques. L’encéphale extériorisé et anormal est adhérent au placenta, fixé lors des mouvements fœtaux (fig. 8.7). Dans cette forme, on retrouve une dysmorphie faciale complexe à type de fentes faciales asymétriques et obliques, d’hypertélorisme, d’anomalie de l’ensellure nasale, de micro- ou anophtalmie unilatérale ;
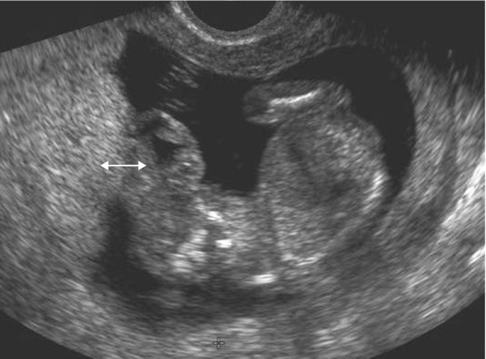
L’encéphale extériorisé est accolé au placenta (↔ ). L’étude en temps réel montre que la tête reste fixée au placenta alors que le reste de l’embryon est mobile.
 le monstre acardiaque–acéphale, malformation exceptionnelle pouvant compliquer une grossesse gémellaire monochoriale (fig. 8.8, et voir fig. 18.35 à 18.37) ;
le monstre acardiaque–acéphale, malformation exceptionnelle pouvant compliquer une grossesse gémellaire monochoriale (fig. 8.8, et voir fig. 18.35 à 18.37) ;
Grossesse gémellaire monochoriale. Coupe frontale du jumeau acardiaque dont on distingue quelques éléments rachidiens lombaires et thoraco-abdominaux (→) et qui est entouré d’un œdème sous-cutané important (∗). Le pôle céphalique est absent.
 le limb body wall complex, forme majeure d’une maladie des brides amniotiques, associant exencéphalie, rachischisis, cœlosomie et amputations de membres (fig. 8.9) ;
le limb body wall complex, forme majeure d’une maladie des brides amniotiques, associant exencéphalie, rachischisis, cœlosomie et amputations de membres (fig. 8.9) ;
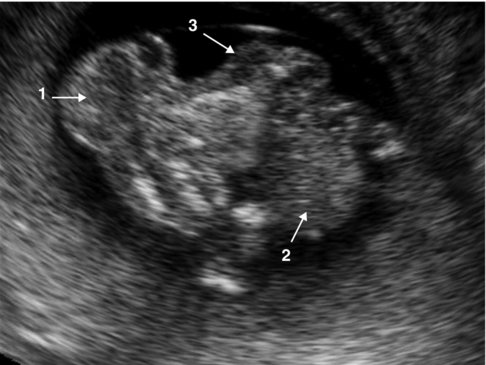
Coupe sagittale de l’embryon qui est polymalformé avec au minimum une exencéphalie (1), une cœlosomie (2) et un rachischisis (3).
 la dysgénésie tubulaire rénale (autosomique récessive) associant hypocalvaria, microcéphalie et retard de croissance, dysmorphie faciale et déformation des membres secondaires à l’anamnios retrouvé en fin de 2e trimestre (séquence de Potter). Les reins gardent un aspect échographique habituel ;
la dysgénésie tubulaire rénale (autosomique récessive) associant hypocalvaria, microcéphalie et retard de croissance, dysmorphie faciale et déformation des membres secondaires à l’anamnios retrouvé en fin de 2e trimestre (séquence de Potter). Les reins gardent un aspect échographique habituel ;
 l’aplasie cutanée congénitale où il existe une acalvaria partielle avec un encéphale sous-jacent et la peau en regard ± normaux (rendant le diagnostic échographique tributaire de la position de la tête fœtale) jusqu’au syndrome d’Adams-Oliver associant, à des degrés variables, acalvaria partielle, aplasie cutanée du vertex, malformations oculaires à type de microphtalmie ou de cataracte, malformations des membres et des extrémités, cardiopathie et sclérose hépato-portale.
l’aplasie cutanée congénitale où il existe une acalvaria partielle avec un encéphale sous-jacent et la peau en regard ± normaux (rendant le diagnostic échographique tributaire de la position de la tête fœtale) jusqu’au syndrome d’Adams-Oliver associant, à des degrés variables, acalvaria partielle, aplasie cutanée du vertex, malformations oculaires à type de microphtalmie ou de cataracte, malformations des membres et des extrémités, cardiopathie et sclérose hépato-portale.
Céphalocèle
Hernie du contenu encéphalique à travers une brèche de la voûte ou de la base du crâne. Elle se rencontre dans 1/5000 naissances et se caractérise par un defect de la voûte avec protrusion dans le liquide amniotique d’une poche méningée ne contenant que du liquide cérébrospinal (méningocèle), mais pouvant ne contenir que du tissu cérébral (encéphalocèle). Si les deux sont présents on parle de méningo-encéphalocèle.
Elle peut être petite – céphalocèle atrétique (fig. 8.10 et 8.11) – ou d’un volume variable avec une composante plus ou moins importante de tissu cérébral et une désorganisation des structures encéphaliques sous-jacentes. Elle peut être localisée au niveau de la base ou de la voûte, sur la ligne médiane, en région frontale et surtout occipitale (75 % des cas) ou latéralement. Le pronostic dépend de la localisation, du volume et du type de céphalocèle. Une encéphalocèle est habituellement de mauvais pronostic, ce qui n’est pas toujours le cas pour une méningocèle isolée notamment si elle est antérieure et de petit volume (inférieure à 20 mm de diamètre). Il peut néanmoins exister des troubles endocriniens associés quel que soit le volume de la méningocèle.
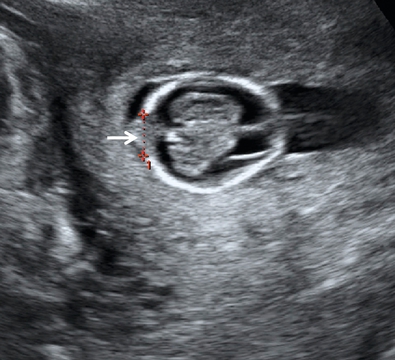
Coupe transversale du pôle céphalique. Les plexus choroïdes et la scissure interhémisphérique ont un aspect normal. On note en revanche une petite solution de continuité à la partie occipitale de la voûte crânienne correspondant à une céphalocèle (→).
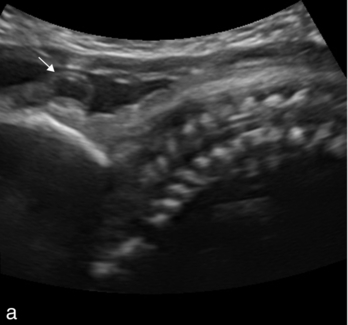
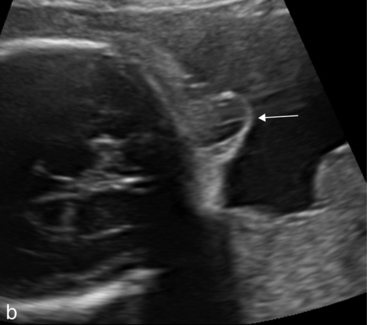
a. Coupe sagittale de la base du crâne et des premières vertèbres cervicales. Petite tumeur hypo-échogène de 1 cm de diamètre située au niveau occipital (→).
b. Coupe transversale inclinée vers le bas du pôle céphalique. La tumeur extériorisée, hypo-échogène, est parsemée de travées linéaires correspondant à des structures nerveuses (→). Il s’agit donc d’une encéphalocèle et non d’une méningocèle.
En échographie, l’aspect découle de la taille du defect crânien et du contenu de la hernie : celui-ci est anéchogène s’il est limité aux méninges et au liquide cérébrospinal (fig. 8.12), mixte, hétérogène, cérébriforme si des structures encéphaliques sont associées (fig. 8.13).
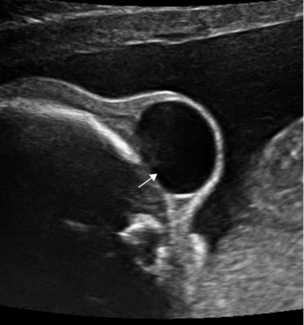
Hernie méningée, au contenu liquidien (anéchogène) s’extériorisant au travers d’un cranioschisis occipital (→).
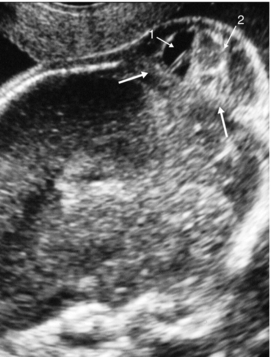
Coupe sagittale du pôle céphalique. Large cranioschisis postérieur (→) laissant s’extérioriser des éléments méningés (1) et encéphaliques (2).
Dans les formes occipitales, la fosse cérébrale postérieure est anormale avec une ou deux cornes occipitales attirées vers le cranioschisis (fig. 8.14). Une ventriculomégalie est retrouvée trois fois sur quatre. En revanche, le cervelet est rarement présent dans une céphalocèle occipitale.
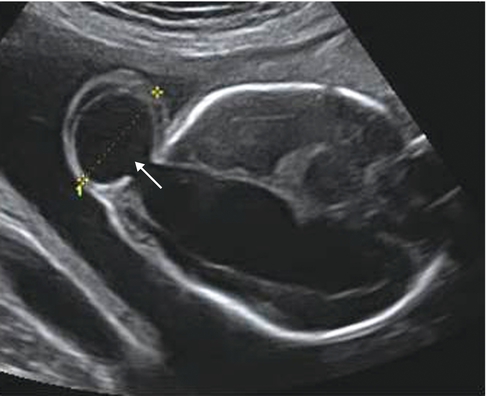
Coupe transversale du pôle céphalique. On individualise une ventriculomégalie unilatérale avec extériorisation de la corne occipitale au travers d’un petit cranioschisis (→).
Les formes frontales sont associées à une fente labiale, labiopalatine ou, dans les formes majeures, à une fente faciale responsable d’un hypertélorisme.
 le syndrome de Meckel-Gruber (autosomique récessif) comportant une céphalocèle occipitale (80 %), une dysplasie rénale multikystique (100 %), une polydactylie post-axiale et un anamnios (voir chap. 14) ;
le syndrome de Meckel-Gruber (autosomique récessif) comportant une céphalocèle occipitale (80 %), une dysplasie rénale multikystique (100 %), une polydactylie post-axiale et un anamnios (voir chap. 14) ;
 le syndrome de Joubert, la dysraphie tectocérébelleuse, le syndrome de Walker-Warburg… (voir plus loin) ;
le syndrome de Joubert, la dysraphie tectocérébelleuse, le syndrome de Walker-Warburg… (voir plus loin) ;
 l’iniencéphalie associant une céphalocèle occipitale et un rachischisis des premières vertèbres cervicales responsable d’une hyperextension du cou (voir fig. 10.22).
l’iniencéphalie associant une céphalocèle occipitale et un rachischisis des premières vertèbres cervicales responsable d’une hyperextension du cou (voir fig. 10.22).
Le diagnostic différentiel est représenté par l’hygroma cervical postérieur dans sa forme pseudo-solide où les septa des logettes sont épais. La fosse cérébrale postérieure est normale et il s’y associe parfois une anasarque fœtale (voir chap. 9). Il est plus difficile d’éliminer une anomalie de la voûte non communicante avec les méninges ou l’encéphale sous-jacent.
Image d’addition de la voûte
Hémangiome sous-cutané
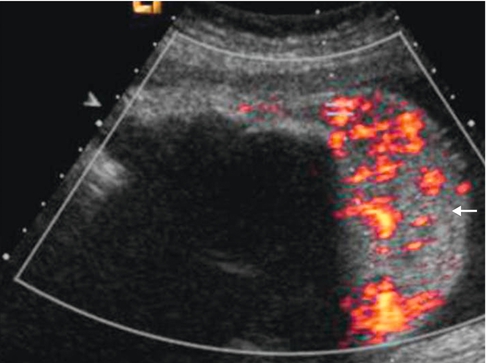
D’un volume variable, il peut être imposant et s’étendre à la face et au thorax, isolé ou associé à des malformations encéphaliques diverses évoquant un syndrome neurocutané. Il peut régresser progressivement et rapidement en post-natal. En échographie, on note une masse hypo-échogène, finement échogène, ou globalement échogène, aux contours d’autant plus bosselés qu’elle est volumineuse, bien individualisée parfois grâce au Doppler couleur qui retrouve plutôt un flux lent (fig. 8.15).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


