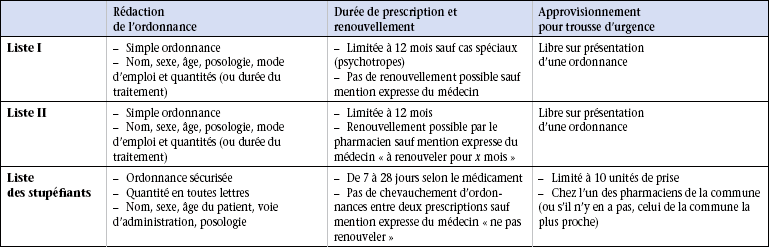
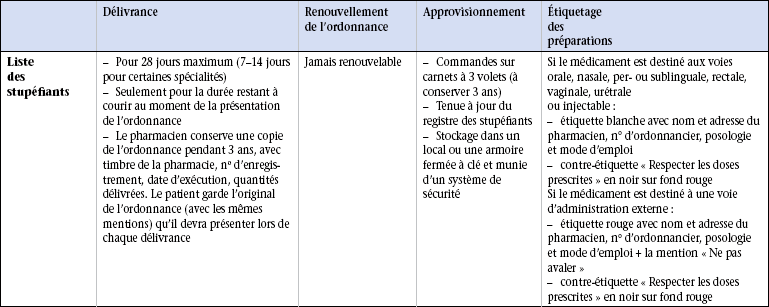
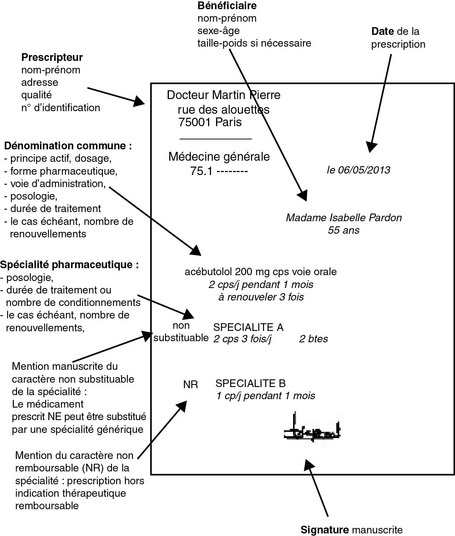
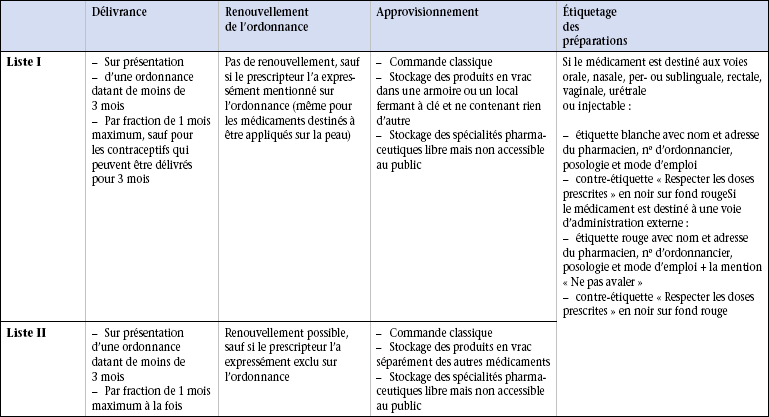
19 Aucun médicament n’est inoffensif. Le Code de la santé publique et de la population réglemente les médicaments dont l’administration peut entraîner des effets toxiques. Le législateur a ainsi pris des mesures permettant de contrôler la délivrance des médicaments à risque pour le patient. Les tableaux 19.1 à 19.3 soulignent les points fondamentaux de cette réglementation et l’étiquetage des préparations. Tableau 19.2 Points essentiels de la délivrance des médicaments des listes I et II par le pharmacien Les stupéfiants sont des médicaments donnant lieu à des abus toxicomaniaques qui engendrent une dépendance physique ou psychique. Ils sont regroupés dans une liste particulière et leur délivrance est soumise à des règles très strictes : l’ordonnance du médecin est sécurisée et comporte un filigrane ainsi qu’un numéro de lot et un carré de microlettres interdisant sa falsification. Selon les médicaments prescrits, la quantité délivrée est limitée à 7, 14 ou 28 jours de traitement. Toute ordonnance doit mentionner (figure 19.1) : • le nom, l’adresse et le numéro de téléphone du prescripteur ; • la date de rédaction de l’ordonnance ; • le nom, le prénom, l’âge et le sexe du patient ; • le nom du médicament ou la dénomination commune internationale (DCI) du principe actif dans la prescription de générique ; • la forme du médicament et le dosage (gélules, comprimés, suppositoires, etc.) ; • la posologie et le mode d’emploi ; • la durée du traitement avec éventuellement la mention du nombre de renouvellements de l’ordonnance ; • le nom du médicament et sa posologie écrite en chiffres ; • la quantité prescrite ou la durée du traitement ; • éventuellement le nombre de renouvellements ; • si le prescripteur veut dépasser pour un médicament la posologie maximale fixée en une fois et en 24 heures, il doit porter sur l’ordonnance la mention « je dis telle dose » ; • l’ordonnance doit dater de moins de 3 mois pour la délivrance des médicaments. Les caractéristiques de prescription sont présentées dans le tableau 19.1. Un médicament générique est la copie d’un médicament original, appelé princeps, ne bénéficiant plus d’une exclusivité commerciale car son brevet est arrivé à terme, 20 ans après son dépôt, et tombe donc dans le domaine public. Cette spécialité peut alors être copiée par d’autres laboratoires que le laboratoire d’origine. Les excipients utilisés dans la fabrication du générique sont différents de ceux utilisés pour le princeps, ce qui peut entraîner des modifications du métabolisme du principe actif dans l’organisme et donc des modifications de l’effet thérapeutique. Un médicament générique doit obtenir une autorisation de mise sur le marché (AMM) qui ne nécessite qu’un test de bioéquivalence, réalisé sur des volontaires sains, dont le but est de s’assurer que les caractéristiques pharmacocinétiques (devenir du médicament dans l’organisme en fonction du temps et de la dose administrée) du générique sont voisines de celles du médicament original. Les autorités de santé admettent une différence inférieure à 20 % du comportement pharmacocinétique du générique par rapport au médicament princeps. On dit qu’il y a alors bioéquivalence entre les deux produits. Outre ce test fondamental de bioéquivalence, le contrôle de la composition chimique et des impuretés du générique est effectué exactement comme pour le princeps.
Réglementation des médicaments listés et des stupéfiants
Classification des médicaments à risque pour la santé
Stupéfiants
Règles générales de rédaction d’une ordonnance
Listes I et II des médicaments
Médicaments génériques et droit de substitution
Principe de bioéquivalence
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


19: Réglementation des médicaments listés et des stupéfiants