L’échographie morphologique des membres fœtaux reste un exercice délicat, malgré les différentes techniques à notre disposition, y compris le mode volumique. Une étude française de 1999 annonçait une sensibilité du dépistage des anomalies des membres de l’ordre de 70 % pour les anomalies majeures et de l’ordre de 20 % pour les anomalies dites mineures des extrémités. Le diagnostic « exact » anténatal des pathologies osseuses dépistées est d’environ 60 %, mais la gravité de celles-ci est le plus souvent correctement évaluée. La période d’exploration optimale se situe entre 12 SA, notamment par voie endovaginale (voir fig. 4.11, 4.12 et 4.31) et 26 SA (voir chap. 6, Membres et extrémités et fig. 6.109 à 6.120). La morphogenèse des membres se déroule entre la 7e et la 10e semaine. Le membre supérieur est constamment en avance de quelques jours sur le membre inférieur. Par échographie abdominale, les os longs des membres deviennent visibles à partir de la 10e semaine d’aménorrhée. Les petits os des extrémités ne le sont qu’à la 14e semaine. Par échographie endovaginale, 1 à 4 semaines peuvent être gagnées. Les membres supérieurs deviennent nettement visibles à la 8e semaine. À la 10e semaine, la longueur totale des deux membres est visible. Le pouce en opposition est visible à la 9e semaine. La conjonction de la voie vaginale, de la haute fréquence, du mode volumique – et de bonnes conditions d’examen – peut produire des images d’une très grande précision dès 12–14 SA, et ce créneau diagnostique n’est sans doute pas assez utilisé (fig. 15.1a). Les pathologies seront décrites ici par type en fonction du signe d’appel principal : Ils font l’objet d’une inspection complète et d’une biométrie plus ou moins exhaustive selon les cas. Les insuffisances biométriques que l’on rencontre dans le cadre des dysplasies osseuses (nanisme) même modérées, sont souvent caricaturales, évidentes au premier coup d’œil et sans commune mesure avec celles d’un RCIU : la longueur se situe à 5, 10 ou 20 mm au-dessous du 3e percentile. L’analyse de la forme des os peut conduire à la découverte de courbures ou de fractures, uniques ou multiples. Il est plus délicat d’apprécier le degré de minéralisation du squelette, sauf au niveau de la boîte crânienne. L’observation des mouvements des différents segments de membres en particulier de flexion et d’extension, constitue l’approche fonctionnelle et concourt à évoquer ou infirmer une arthrogrypose ou une maladie des brides amniotiques par exemple. L’aspect « gros ventre + petit thorax » (fig. 15.1b) est un élément sémiologique souvent frappant. Le périmètre thoracique peut être mesuré mais l’appréciation subjective est largement suffisante pour évoquer le thorax étroit (tronc en « bouchon de champagne »). Le degré d’hypoplasie pulmonaire peut être apprécié de façon indirecte : L’hypoplasie pulmonaire peut aussi être évaluée de façon plus spécifique par la mesure de la surface pulmonaire ou le calcul du LHR (comme cela se pratique dans l’évaluation pronostique d’une hernie de coupole diaphragmatique) (voir fig. 7.17, 7.18 et 11.10). Le mode 3D permet théoriquement une mesure directe du volume pulmonaire mais il s’agit d’une biométrie difficile nécessitant un échographiste entraîné et qui peut être remplacée avantageusement par l’IRM pulmonaire. De plus, le volume pulmonaire ne préjuge pas automatiquement de la capacité fonctionnelle. Outre les défauts de fermeture, elle peut être le siège de duplications, d’élargissements ou d’anomalies de courbure isolées ou liées à la présence d’hémi-vertèbres (voir chap. 10). Les anomalies de nombre des vertèbres et leur degré de minéralisation sont difficiles à mettre en évidence : la radiologie classique, ou le scanner fœtal, et l’IRM seront très utiles. Le mode volumique peut aussi être précieux, en particulier le mode 4D « osseux » mais, comme souvent en 3D-4D, il faut de bonnes conditions et un fœtus coopérant (voir fig. 6.54). L’observation de la forme du crâne permet de décrire le front (haut, en tour, dans le syndrome d’Apert ou bombant dans les chondrodysplasies et certaines hydrocéphalies ou fuyant dans le syndrome de Pierre Robin, par exemple), les bosses frontales (proéminentes dans l’achondroplasie), l’écaille occipitale (plate dans les craniosténoses). Le degré de minéralisation est mieux estimé au niveau du pôle céphalique que partout ailleurs (transparence aux ultrasons, déformation sous la sonde). Les formes terminales (ou distales) sont plus volontiers d’origine génétique lorsqu’elles sont bilatérales et sont plus souvent associées à d’autres pathologies quand elles touchent les membres inférieurs plutôt que les membres supérieurs. L’amélie dont la fréquence est estimée à 0,15/10 000 naissances correspond à l’absence complète d’un ou de plusieurs membres : Les publications ont fleuri sur la responsabilité des choriocentèses dans la genèse des amputations transverses terminales : pour que cette étiologie soit retenue, il faut que la biopsie de trophoblaste ait été pratiquée trop précocement, avant la 10e semaine. La maladie des brides amniotiques peut amputer les membres transversalement de façon extrêmement variable (fig. 15.2, et voir fig. 5.10). En faveur de cette étiologie, on retient l’œdème des membres atteints et la présence d’un anneau de striction. Il faut penser au Doppler couleur pour évaluer le degré de striction par absence ou diminution du flux vasculaire sous ce niveau. Le risque de récidive est nul. Des cas de grossesses gémellaires avec mort in utero du co-jumeau et amputation de membres secondaires à une disruption vasculaire (collapsus circulatoire) chez le jumeau survivant ont été décrits. Le tableau mime celui des brides amniotiques alors qu’elles sont absentes. L’hémimélie correspond à l’absence distale des membres, en dessous du coude ou du genou. Elle peut être partielle ou totale et appartenir à des syndromes comme l’aglossie–adactylie (rétrognathisme, aglossie, hydramnios, amputation de membres à des degrés divers). Ces associations malformatives sont sporadiques. Parmi les agénésies des extrémités (fig. 15.3), celles des mains (acheirie) ou des pieds (apodie) sont habituellement isolées. L’exceptionnelle association des deux (acheiropodie), a été décrite au Brésil dans le cadre d’un syndrome récessif autosomique. Les adactylies (absence de doigts et d’orteils) sont d’une grande variété et d’un grand polymorphisme (fig. 15.4), isolées ou entrant dans des syndromes divers incluant essentiellement une pathologie buccale tel le syndrome hypoglossie–hypodactylie (syndrome d’Hanhart) qui associe rétrognathisme, ankyloglossie responsable de troubles de la déglutition avec hydramnios et amputation transverse terminale de sévérité variable. L’ectrodactylie correspond à l’absence d’un ou de plusieurs doigts centraux. Il s’agit essentiellement de formes familiales, isolées à transmission dominante autosomique. Sans notion d’antécédents familiaux, ces formes isolées sont peu souvent diagnostiquées par échographie. Les phocomélies sont des agénésies transverses d’un segment moyen ou proximal, préservant la main et le pied (fig. 15.5). La thalidomide est à l’origine des phocomélies complètes mais il existe des formes isolées sporadiques. Ces agénésies peuvent s’inscrire dans quelques syndromes rares et complexes : Au niveau des membres supérieurs, elles sont soit radiales, soit cubitales. Celles des membres inférieurs sont externes, touchant le péroné (fig. 15.6), ou internes, touchant le tibia. Dans tous les cas, les agénésies longitudinales sont associées à des anomalies de position des extrémités qui seront abordées plus loin dans ce chapitre. Elles sont longitudinales, d’importance et de siège variable, sur un ou plusieurs membres. Elles peuvent intégrer différents syndromes. La brièveté des membres regroupe des pathologies très diverses. Les ostéochondrodysplasies et, dans une moindre mesure, les anomalies chromosomiques constituent l’essentiel du problème des membres courts. La classification des chondrodysplasies est ardue en raison d’une méconnaissance de leur pathogénie et la terminologie adoptée est fonction de la description sémiologique (fig. 15.7 et tableau 15.1) : Tableau 15.1 Classification descritive des nanismes (D : domimant ; R : récessif ; S : sporadique) D’après Spirt BA et al. Prenatal sonographic evaluation of short-limbed dwarfism : an algorithmic approach. RadioGraphics 1990 ; 10(2) : 217–36. Tous les os longs sont mesurés et rapportés à des valeurs standard pour qualifier le type d’atteinte (voir tableaux 7.4, 7.5, 7.26 et 7.27). Dans les nanismes les plus fréquents, le raccourcissement se situe à plus de deux déviations standard (DS) sous les normes, ce qui correspond à une mesure inférieure au 3e percentile de la courbe de référence. Les raccourcissements modérés du fémur et de l’humérus correspondent le plus souvent à une hypotrophie fœtale ou à une trisomie. Certains cas d’achondroplasie n’ont qu’une atteinte modérée au 2e trimestre mais elle s’accentue franchement à partir du 6e mois. La grande majorité des chondrodysplasies présente des disproportions majeures, souvent évidentes à la première inspection et inférieures de plusieurs millimètres (voire centimètres) au 3e percentile.
Pathologie des membres et des extrémités
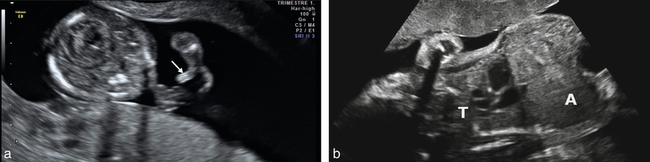
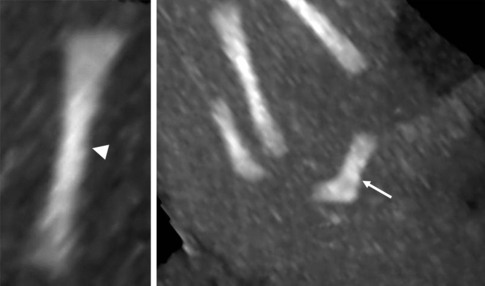
a. Nanisme diastrophique. 13 SA et 2 jours. Diagnostic évoqué précocément par voie vaginale. Les membres sont courts et la position des pouces « faisant de l’auto-stop » (→) est très évocateur.
b. Signe d’appel à 28 SA. Thorax étroit avec aspect gros ventre (A) et petit thorax (T) porte d’entrée vers un diagnostic de chondrodysplasie. Source fig. 15.1a : Dr P. Vaast.
 le membre est amputé transversalement ou longitudinalement : on parle d’agénésie. Les agénésies ont une fréquence moyenne de 5/1000 naissances, soit cinq fois plus que celle de la trisomie 21 dans une population de femmes de 25 à 30 ans. Lorsqu’elles sont transversales, elles sont isolées dans la moitié des cas. Lorsqu’elles sont longitudinales, elles entrent majoritairement dans le cadre d’une pathologie malformative, de pronostic et de prise en charge totalement différents ;
le membre est amputé transversalement ou longitudinalement : on parle d’agénésie. Les agénésies ont une fréquence moyenne de 5/1000 naissances, soit cinq fois plus que celle de la trisomie 21 dans une population de femmes de 25 à 30 ans. Lorsqu’elles sont transversales, elles sont isolées dans la moitié des cas. Lorsqu’elles sont longitudinales, elles entrent majoritairement dans le cadre d’une pathologie malformative, de pronostic et de prise en charge totalement différents ;
 le membre présente une brièveté : la démarche diagnostique est fonction de l’importance de cette brièveté, de l’atteinte d’un ou plusieurs segments, de l’uni- ou bilatéralité, du siège sur un ou plusieurs membres, d’une déformation surajoutée, de la date du diagnostic (précoce dès le 1er ou le 2e trimestre ou tardive en fin de grossesse). À côté d’une petite taille constitutionnelle qui est parfois évoquée, cette rubrique comporte les anomalies chromosomiques, mais aussi certains syndromes et toutes les chondrodysplasies et nanismes au sens large. La sommation des signes oriente l’enquête diagnostique ;
le membre présente une brièveté : la démarche diagnostique est fonction de l’importance de cette brièveté, de l’atteinte d’un ou plusieurs segments, de l’uni- ou bilatéralité, du siège sur un ou plusieurs membres, d’une déformation surajoutée, de la date du diagnostic (précoce dès le 1er ou le 2e trimestre ou tardive en fin de grossesse). À côté d’une petite taille constitutionnelle qui est parfois évoquée, cette rubrique comporte les anomalies chromosomiques, mais aussi certains syndromes et toutes les chondrodysplasies et nanismes au sens large. La sommation des signes oriente l’enquête diagnostique ;
 le membre présente des anomalies de segmentation : polydactylies ou syndactylies. Les premières, le plus souvent isolées, sont volontiers des formes familiales, de transmission dominante autosomique. Les secondes, difficiles d’accès à l’échographie, peuvent éventuellement l’être par le biais des signes d’appel que constituent les anomalies associées ;
le membre présente des anomalies de segmentation : polydactylies ou syndactylies. Les premières, le plus souvent isolées, sont volontiers des formes familiales, de transmission dominante autosomique. Les secondes, difficiles d’accès à l’échographie, peuvent éventuellement l’être par le biais des signes d’appel que constituent les anomalies associées ;
 le membre est malposé : les pieds bots sont les anomalies les plus fréquentes dans ce registre, puis viennent les mains botes. Les pieds varus et les mains en coup de vent sont plus rares. Le caractère uni- ou bilatéral de ces lésions, isolé ou entrant dans un contexte malformatif, portant sur le segment de membre sus-jacent ou touchant une autre sphère, leur confère une implication pronostique différente. L’examen fonctionnel porte sur le ou les membres atteints pour apprécier le degré de fixation de la malposition, et sur l’ensemble du fœtus à la recherche d’un immobilisme ;
le membre est malposé : les pieds bots sont les anomalies les plus fréquentes dans ce registre, puis viennent les mains botes. Les pieds varus et les mains en coup de vent sont plus rares. Le caractère uni- ou bilatéral de ces lésions, isolé ou entrant dans un contexte malformatif, portant sur le segment de membre sus-jacent ou touchant une autre sphère, leur confère une implication pronostique différente. L’examen fonctionnel porte sur le ou les membres atteints pour apprécier le degré de fixation de la malposition, et sur l’ensemble du fœtus à la recherche d’un immobilisme ;
 le membre est déformé sans être raccourci : c’est le problème de l’incurvation des os longs.
le membre est déformé sans être raccourci : c’est le problème de l’incurvation des os longs.
Bilan malformatif
Squelette
Os longs
Thorax
 les normes du rapport périmètre thoracique/périmètre abdominal se situent entre 0,77 et 1,01 ;
les normes du rapport périmètre thoracique/périmètre abdominal se situent entre 0,77 et 1,01 ;
 celles du rapport cardiothoracique établi sur une coupe quatre cavités est d’un tiers et ne doit pas dépasser un demi (fig. 12.8).
celles du rapport cardiothoracique établi sur une coupe quatre cavités est d’un tiers et ne doit pas dépasser un demi (fig. 12.8).
Colonne vertébrale
Crâne
Agénésies et hypoplasies
Agénésies transverses
 unique, elle a plutôt un caractère isolé ;
unique, elle a plutôt un caractère isolé ;
 quadruple, elle est plutôt d’origine médicamenteuse ou infectieuse, par exemple par la varicelle survenue avant la fin du 1er trimestre de gestation.
quadruple, elle est plutôt d’origine médicamenteuse ou infectieuse, par exemple par la varicelle survenue avant la fin du 1er trimestre de gestation.
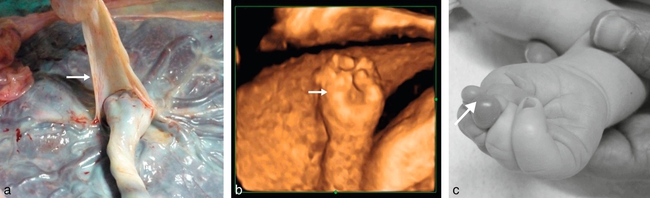
a. Bride amniotique large et épaisse (→).
b. 25 SA. Étude en mode 3D surface. Anomalie de la main (→) qui est rétractée avec syndactylies.
c. ædème des extrémités (→) des trois premiers doigts (autre fœtus).
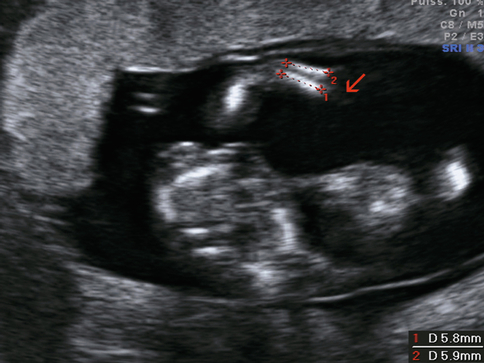
Les deux os de l’avant-bras sont présents mais un peu courts (< 6 mm). La main est absente (→).
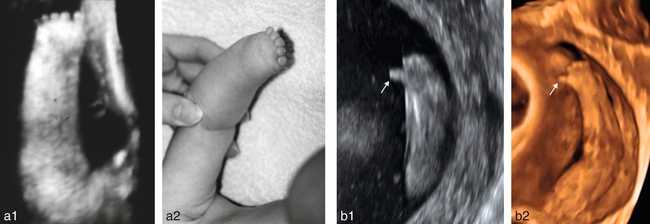
a. Adactylie isolée : bonne corrélation entre l’échographie 3D (a1) et l’aspect à la naissance (a2). b. Adactylie. 21 SA. Étude en mode bidimensionnel (b1) et en mode 3D surface (b2). Le pouce paraît
normal (→) mais les quatre autres doigts sont absents. Le poignet et la paume de la main sont présents. Il s’agissait d’une adactylie sans autre malformation décelable.
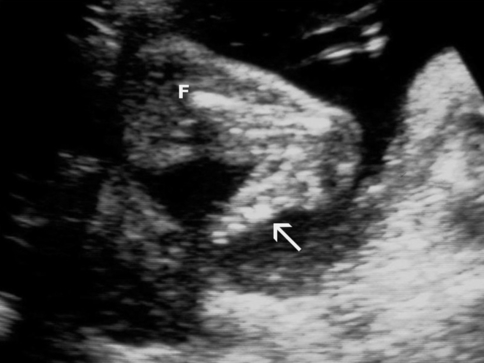
Le pied (→) est directement implanté sur le bas de la cuisse repérée par le fémur (F).
 le syndrome de Roberts (voir tableau 8.1), ou pseudo-thalidomide syndrome, associe un retard de croissance constant et sévère, des anomalies craniofaciales à type de fente labiomaxillaire et de microcéphalie, et des malformations des membres allant de la phocomélie partielle à la tétraphocomélie. Le pronostic est le plus souvent létal en période périnatale secondaire aux malformations cardiaques et rénales. Le retard mental est constant. Secondaire à une mutation du gène ESCO2, la transmission est récessive autosomique ;
le syndrome de Roberts (voir tableau 8.1), ou pseudo-thalidomide syndrome, associe un retard de croissance constant et sévère, des anomalies craniofaciales à type de fente labiomaxillaire et de microcéphalie, et des malformations des membres allant de la phocomélie partielle à la tétraphocomélie. Le pronostic est le plus souvent létal en période périnatale secondaire aux malformations cardiaques et rénales. Le retard mental est constant. Secondaire à une mutation du gène ESCO2, la transmission est récessive autosomique ;
 le syndrome de Cornelia de Lange (voir plus loin et tableau 8.1) peut comporter des phocomélies et une oligodactylie. De survenue habituellement sporadique, il peut être lié à des mutations autosomiques dominantes du gène NIPBL.
le syndrome de Cornelia de Lange (voir plus loin et tableau 8.1) peut comporter des phocomélies et une oligodactylie. De survenue habituellement sporadique, il peut être lié à des mutations autosomiques dominantes du gène NIPBL.
Agénésies longitudinales
Hypoplasies
 Le CHILD syndrome (acronyme pour congenital hemidysplasia ichtyosis limb defect) est une entité exceptionnelle comportant une atteinte unilatérale des extrémités, pouvant aller d’une simple hypoplasie des phalanges jusqu’à l’absence d’un membre, une alopécie et une atteinte cutanée ipsilatérales, un érythème, une hyperkératose. L’atteinte viscérale siège sur les reins sous forme d’une hydronéphrose ou d’une agénésie rénale, ou s’exprime par une cardiopathie. La sex-ratio, de 19 filles pour 1 garçon, fait suspecter une transmission dominante liée à l’X.
Le CHILD syndrome (acronyme pour congenital hemidysplasia ichtyosis limb defect) est une entité exceptionnelle comportant une atteinte unilatérale des extrémités, pouvant aller d’une simple hypoplasie des phalanges jusqu’à l’absence d’un membre, une alopécie et une atteinte cutanée ipsilatérales, un érythème, une hyperkératose. L’atteinte viscérale siège sur les reins sous forme d’une hydronéphrose ou d’une agénésie rénale, ou s’exprime par une cardiopathie. La sex-ratio, de 19 filles pour 1 garçon, fait suspecter une transmission dominante liée à l’X.
 Le syndrome de régression caudale (voir fig. 10.24) se manifeste à des degrés variables, allant de l’agénésie lombo-sacrée à la sirénomélie, séquence malformative atteignant strictement les membres inférieurs et incluant des anomalies viscérales (agénésie rénale, vésicale, rectale avec imperforation anale). Les étiologies sont multiples et mal connues en dehors de la pathologie vasculaire (sirénomélie) et des complications diabétiques (formes partielles du syndrome de régression caudale). Les enfants survivants n’ont pas de problème mental, mais la morbidité au long court est d’origine orthopédique et neurologique avec souffrance vésicorénale.
Le syndrome de régression caudale (voir fig. 10.24) se manifeste à des degrés variables, allant de l’agénésie lombo-sacrée à la sirénomélie, séquence malformative atteignant strictement les membres inférieurs et incluant des anomalies viscérales (agénésie rénale, vésicale, rectale avec imperforation anale). Les étiologies sont multiples et mal connues en dehors de la pathologie vasculaire (sirénomélie) et des complications diabétiques (formes partielles du syndrome de régression caudale). Les enfants survivants n’ont pas de problème mental, mais la morbidité au long court est d’origine orthopédique et neurologique avec souffrance vésicorénale.
 L’hypoplasie fémorale, totale ou partielle, peut être associée à un faciès particulier, petit nez avec hypoplasie des ailes du nez, philtrum long, rétrognathisme et fente palatine. L’aspect de la face est un important élément d’orientation. Un diabète maternel est souvent retrouvé. La majorité des cas sont sporadiques. Il en est de même pour l’hypoplasie humérale, de nature strictement isolée. L’hypoplasie fémorale peut aussi être isolée, unilatérale dans 90 % des cas et relativement fréquente (1/800 naissances). Le diagnostic ne sera posé par échographie qu’en cas d’asymétrie évidente de taille des fémurs. Elle résulte d’une anomalie de développement de la partie sous-trochantérienne du fémur entraînant un raccourcissement à des degrés variables de l’extrémité supérieure du fémur. La tête fémorale et l’acétabulum peuvent également être anormaux rendant complexe la prise en charge orthopédique.
L’hypoplasie fémorale, totale ou partielle, peut être associée à un faciès particulier, petit nez avec hypoplasie des ailes du nez, philtrum long, rétrognathisme et fente palatine. L’aspect de la face est un important élément d’orientation. Un diabète maternel est souvent retrouvé. La majorité des cas sont sporadiques. Il en est de même pour l’hypoplasie humérale, de nature strictement isolée. L’hypoplasie fémorale peut aussi être isolée, unilatérale dans 90 % des cas et relativement fréquente (1/800 naissances). Le diagnostic ne sera posé par échographie qu’en cas d’asymétrie évidente de taille des fémurs. Elle résulte d’une anomalie de développement de la partie sous-trochantérienne du fémur entraînant un raccourcissement à des degrés variables de l’extrémité supérieure du fémur. La tête fémorale et l’acétabulum peuvent également être anormaux rendant complexe la prise en charge orthopédique.
Brièveté des membres
 L’atteinte est bilatérale et symétrique, portant sur plusieurs segments de membres, à la fois au niveau des membres supérieurs et des membres inférieurs : les ostéochondrodysplasies ou dysplasies du squelette forment l’essentiel de ce chapitre.
L’atteinte est bilatérale et symétrique, portant sur plusieurs segments de membres, à la fois au niveau des membres supérieurs et des membres inférieurs : les ostéochondrodysplasies ou dysplasies du squelette forment l’essentiel de ce chapitre.
 L’atteinte est bilatérale et porte sur un seul segment de membre, fémur le plus souvent (et/ou humérus) : les anomalies chromosomiques sont évoquées en priorité si les mesures sont faibles précocement dans la grossesse. La croissance osseuse peut également être altérée dès le milieu du 2e trimestre en cas de RCIU précoce d’origine vasculaire. La prise en compte d’une participation constitutionnelle est à évoquer en présence d’une biométrie un peu faible et isolée de survenue plus tardive avant d’évoquer une éventuelle pathologie squelettique.
L’atteinte est bilatérale et porte sur un seul segment de membre, fémur le plus souvent (et/ou humérus) : les anomalies chromosomiques sont évoquées en priorité si les mesures sont faibles précocement dans la grossesse. La croissance osseuse peut également être altérée dès le milieu du 2e trimestre en cas de RCIU précoce d’origine vasculaire. La prise en compte d’une participation constitutionnelle est à évoquer en présence d’une biométrie un peu faible et isolée de survenue plus tardive avant d’évoquer une éventuelle pathologie squelettique.
 L’atteinte touche l’un ou l’autre segment de membre de façon anarchique, réalisant des asymétries de croissance : il s’agit essentiellement du syndrome de Klippel-Trenaunay.
L’atteinte touche l’un ou l’autre segment de membre de façon anarchique, réalisant des asymétries de croissance : il s’agit essentiellement du syndrome de Klippel-Trenaunay.
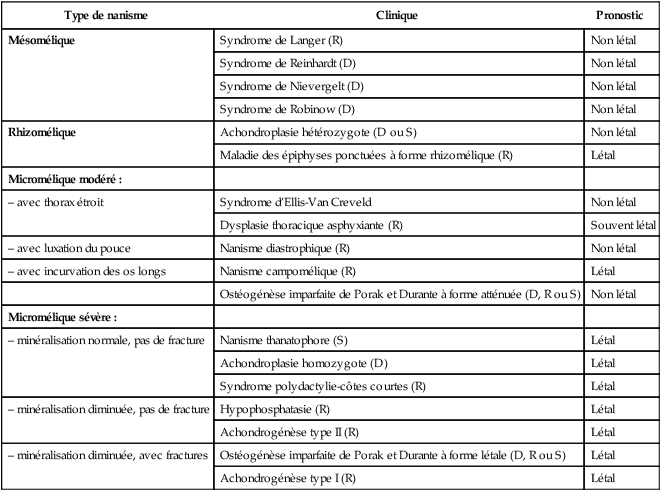
Dysplasies du squelette ou maladies osseuses constitutionnelles
Classifications
 les ostéochondrodysplasies qui réalisent des anomalies de taille (nanisme) et/ou de forme des os et/ou des cartilages avec une fréquence de 2,3 à 4,7 pour 10 000 naissances ;
les ostéochondrodysplasies qui réalisent des anomalies de taille (nanisme) et/ou de forme des os et/ou des cartilages avec une fréquence de 2,3 à 4,7 pour 10 000 naissances ;
 les dysostoses qui sont des malformations d’un os isolé, éventuellement associées à d’autres malformations ;
les dysostoses qui sont des malformations d’un os isolé, éventuellement associées à d’autres malformations ;
 les ostéolyses idiopathiques ;
les ostéolyses idiopathiques ;
 les anomalies chromosomiques (voir chap. 16) sachant que les malformations du squelette et des membres sont le signe d’appel d’anomalies chromosomiques dans environ 7 % des cas, en particulier pour les trisomies 13 et 18. Le fémur court (mesure inférieure au 3e percentile) et un rapport fémur/pied inférieur ou égal à 0,84, font suspecter une trisomie 21. En réalité, l’humérus est plus souvent court que le fémur en cas de trisomie 21 et sa mesure devrait être systématique ;
les anomalies chromosomiques (voir chap. 16) sachant que les malformations du squelette et des membres sont le signe d’appel d’anomalies chromosomiques dans environ 7 % des cas, en particulier pour les trisomies 13 et 18. Le fémur court (mesure inférieure au 3e percentile) et un rapport fémur/pied inférieur ou égal à 0,84, font suspecter une trisomie 21. En réalité, l’humérus est plus souvent court que le fémur en cas de trisomie 21 et sa mesure devrait être systématique ;
Type de nanisme
Clinique
Pronostic
Mésomélique
Syndrome de Langer (R)
Non létal
Syndrome de Reinhardt (D)
Non létal
Syndrome de Nievergelt (D)
Non létal
Syndrome de Robinow (D)
Non létal
Rhizomélique
Achondroplasie hétérozygote (D ou S)
Non létal
Maladie des épiphyses ponctuées à forme rhizomélique (R)
Létal
Micromélique modéré :
– avec thorax étroit
Syndrome d’Ellis-Van Creveld
Non létal
Dysplasie thoracique asphyxiante (R)
Souvent létal
– avec luxation du pouce
Nanisme diastrophique (R)
Non létal
– avec incurvation des os longs
Nanisme campomélique (R)
Létal
Ostéogénèse imparfaite de Porak et Durante à forme atténuée (D, R ou S)
Non létal
Micromélique sévère :
– minéralisation normale, pas de fracture
Nanisme thanatophore (S)
Létal
Achondroplasie homozygote (D)
Létal
Syndrome polydactylie-côtes courtes (R)
Létal
– minéralisation diminuée, pas de fracture
Hypophosphatasie (R)
Létal
Achondrogénèse type II (R)
Létal
– minéralisation diminuée, avec fractures
Ostéogénèse imparfaite de Porak et Durante à forme létale (D, R ou S)
Létal
Achondrogénèse type I (R)
Létal
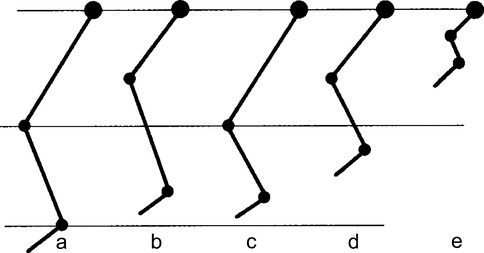
Proportions normales du membre inférieur (a). Nanismes : RHIZOmélique (b) ; MÉSOmélique (c) ; MICROmélique modéré (d) ; MICROmélique sévère (e).
 nanisme rhizomélique, lorsque l’atteinte est proximale ;
nanisme rhizomélique, lorsque l’atteinte est proximale ;
 nanisme mésomélique, lorsqu’elle touche le segment moyen ;
nanisme mésomélique, lorsqu’elle touche le segment moyen ;
 nanisme acromélique, lorsqu’elle touche le segment distal (mains et pieds) ;
nanisme acromélique, lorsqu’elle touche le segment distal (mains et pieds) ;
 nanisme micromélique, lorsque tous les segments de membres sont concernés.
nanisme micromélique, lorsque tous les segments de membres sont concernés.
Explorations échographiques, radiologiques et génétiques
Étude biométrique
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


15: Pathologie des membres et des extrémités