Vers la fin du 1er mois, la délimitation de l’embryon (voir fig. 10.1 et 10.2) correspond à la fermeture du corps par un double phénomène : soulèvement de la plaque en raison d’un développement dorsal beaucoup plus rapide (système nerveux) et formation d’un pli périphérique qui, relativement, se resserre comme le collet d’une bourse. On passe ainsi du petit disque tridermique plat à la structure typique en « haricot ». Le disque repose sur la cavité du lécithocèle que la fermeture du collet (futur ombilic) va venir étrangler et diviser en deux parties : Ces deux parties sont reliées par le canal vitellin qui s’allonge au fur et à mesure que la vésicule ombilicale s’éloigne de l’embryon. Cette vésicule régresse précocement et disparaît aux environs du 3e mois. Vers 7–8 SA, le cordon ombilical se forme par fusion du pédicule de fixation (contenant l’allantoïde, la veine ombilicale et les artères ombilicales) et du canal vitellin. Le développement de l’anse iléale primitive est caractérisé par son allongement extrêmement rapide qui l’oblige à se développer en dehors de l’abdomen en faisant hernie dans le cœlome extra-embryonnaire du cordon à travers l’ombilic, c’est la classique hernie ombilicale physiologique qui est visible en échographie de la 8e à la 11–12e SA (28 % à 8 SA, 72 % à 9 SA, 100 % à 10 SA) (fig. 13.2, et voir fig. 4.9, 4.10 et 4.19). L’élargissement de la cavité abdominale associée à une diminution de la taille relative du foie et des reins permet une réintégration des anses intestinales : c’est le phénomène de réduction. Après 12 SA révolues, en échographie, on ne doit plus mettre en évidence de hernie ombilicale. Un balayage transversal descendant, suivant les contours antérieurs de l’abdomen, est souvent suffisant pour repérer un défaut de fermeture, le diagnostic en est évidemment facilité par la position du dos en arrière. Il faut toujours bien identifier l’implantation abdominale du cordon. Cependant, la coupe idéale (mais pas toujours réalisable) pour l’étude de la paroi abdominale est une coupe sagittale se glissant entre les cuisses, permettant d’identifier de haut en bas la paroi thoracique puis abdominale, l’ombilic, la vessie et le sexe (voir fig. 6.80 et 6.81). Il s’agit d’un diagnostic échographique précoce, dans la majorité des cas dès le 1er trimestre. La surveillance doit être mensuelle puis plus rapprochée en fin de grossesse à la recherche de malformations associées et de complications. Dans les cas « simples », à l’exception de l’exstrophie vésicale, le pronostic postopératoire est généralement bon. Anatomiquement, le defect pariétal antérieur est appelé célosomie, ou cœlosomie (synonymes), lorsqu’il résulte d’un trouble de la morphogenèse. Les célosomies traduisent un échec plus ou moins étendu de la délimitation par arrêt de progression du plissement (les embryologistes parlent d’« avortement » d’un pli) : En raison de leur mécanisme, les célosomies moyennes et supérieures sont caractérisées par la persistance d’une membrane amniotique pour recouvrir le defect. Dans l’exstrophie vésicale et cloacale, la vessie ou les éléments du cloaque viennent occuper le défaut de fermeture et s’ouvrent directement dans la cavité amniotique. Le laparoschisis résulte d’un accident beaucoup plus tardif, bien après la délimitation, et il se présente comme une aplasie ou une nécrose localisée de la paroi, probablement d’origine vasculaire. Le defect pariétal est complet, il n’y a pas de sac herniaire (fig. 13.3b). Pour les embryologistes, le laparoschisis n’appartient donc pas strictement au groupe des célosomies. L’omphalocèle (fig. 13.3a) comporte un defect pariétal important, alors que la hernie ombilicale (fig. 13.3c) correspond à un défaut de réintégration sans élargissement important de l’orifice. La hernie n’est pas toujours visible in utero mais on la craint toujours chez le nouveau-né – même si le phénomène est rare –, ce qui conduit à toujours clamper le cordon à quelques centimètres de son insertion par crainte de pincer une anse intestinale. Il s’agit d’un défaut de fermeture ventrale ou célosomie moyenne, concernant l’orifice ombilical lui-même, donc de siège central (fig. 13.4). Sa fréquence est environ de 1/4000 naissances (1/1000 grossesses en fin de 1er trimestre) et augmente avec l’âge maternel. L’omphalocèle est plus fréquente chez les garçons que chez les filles (5/1). Il n’existe pas de cause tératogène connue. Les anomalies associées avec ou sans pathologies chromosomiques sont fréquentes. Survenant très tôt dans la vie embryonnaire, elle associe une absence de réintégration de l’anse intestinale primitive et souvent une protrusion du foie. Ces malformations viscérales peuvent se regrouper en syndromes plus ou moins complexes : – une omphalocèle sus-ombilicale (pouvant contenir l’estomac), – une ectopie cardiaque partielle (le cœur est engagé dans l’omphalocèle), – une malformation sternale inférieure (agénésie ou fente), – une ouverture diaphragmatique (entraînant une hernie diaphragmatique si elle est importante), – une ouverture du péricarde apical, – l’association est possible avec d’autres malformations (fente labiale ou labiopalatine, encéphalocèle, exencéphalie, sirénomélie, cardiopathie notamment tétralogie de Fallot). Une aberration chromosomique peut être associée. Le pronostic est évidemment désastreux ; – l’ectopie cardiaque (éviscération d’un cœur généralement malformé au travers d’une ouverture pariétale, sternale et/ou péricardique), souvent associée à une omphalocèle et à des anomalies craniofaciales et chromosomique type trisomie 18, – l’agénésie sternale isolée : la paroi antérieure du thorax est pulsatile, synchrone des battements cardiaques. Le cœur peut paraître trop antérieur mais la paroi du thorax est intacte. Il peut s’y associer un hémangiome pariétal masquant le diagnostic ; La réalisation d’un caryotype doit être systématique, du fait de la fréquence (environ 30 %) des anomalies chromosomiques associées (trisomie 18 et 13, plus rarement 21, triploïdie et monosomie X). Cette fréquence est encore augmentée : Le laparoschisis se traduit par une issue de viscères abdominaux au travers d’une aplasie de la paroi abdominale antérieure située le plus souvent au niveau du bord droit de l’ombilic (fig. 13.9a). Sa fréquence varie selon les séries de 1 pour 3000 à 1 pour 10 000 naissances vivantes. Le laparoschisis est beaucoup plus fréquent chez les patientes nulligestes, de moins de 20 ans. Des agents tératogènes sont parfois évoqués (alcool, cocaïne, tabac, pseudo-éphédrine, aspirine, amphétamines, solvants organiques…). Le terme synonyme de gastroschisis est parfois utilisé dans la littérature (surtout en langue anglaise). On parle aussi d’abdominoschisis mais plutôt pour des defects plus larges et plus complexes comme on peut les retrouver dans la maladie amniotique. Les lésions intestinales sont dues à l’irritation produite par le liquide amniotique entraînant un épaississement des anses intestinales herniées (péritonite chimique) qui finissent par s’agglutiner de manière compacte (fig. 13.9b). La striction progressive au niveau du collet (l’intestin grandit plus rapidement que le defect pariétal) peut être responsable d’une anomalie du retour veineux et lymphatique avec œdème, ou d’une ischémie pouvant conduire à une sténose, une atrésie, une nécrose et une perforation intestinale. Ces lésions se développent surtout en fin de grossesse. Le diagnostic échographique peut en être posé tôt et aisément, dès l’échographie de dépistage du 1er trimestre où, sur une coupe sagittale de l’embryon complétée par une coupe transversale de l’abdomen, on met en évidence au niveau ombilical une image d’addition aux contours mal définis, mobile avec le fœtus (fig. 13.10a). Lors de l’étude morphologique au 2e trimestre, on note (fig. 13.10b à d) : Elle fait partie des célosomies inférieures et se traduit par un defect pariétal bas et une ouverture de la vessie : la muqueuse vésicale, plus ou moins végétante, fait saillie à ce niveau et se poursuit directement avec la peau avoisinante. Cette malformation, retrouvée dans 1/30 000 naissances, est plus fréquente chez le garçon, avec des conséquences également plus graves sur le plan urinaire et sexuel. En échographie, le signe essentiel est l’absence permanente de visualisation de la vessie, alors que le haut appareil urinaire est normal ainsi que la quantité de liquide amniotique. La muqueuse vésicale saillante boursoufle de façon irrégulière la région sous-ombilicale qui prend un aspect « fripé » (fig. 13.12a). L’examen Doppler couleur peut aider à localiser la plaque vésicale en montrant les artères ombilicales dans le pelvis.
Pathologie de la paroi et du contenu abdominal
Rappel embryologique (fig. 13.1)
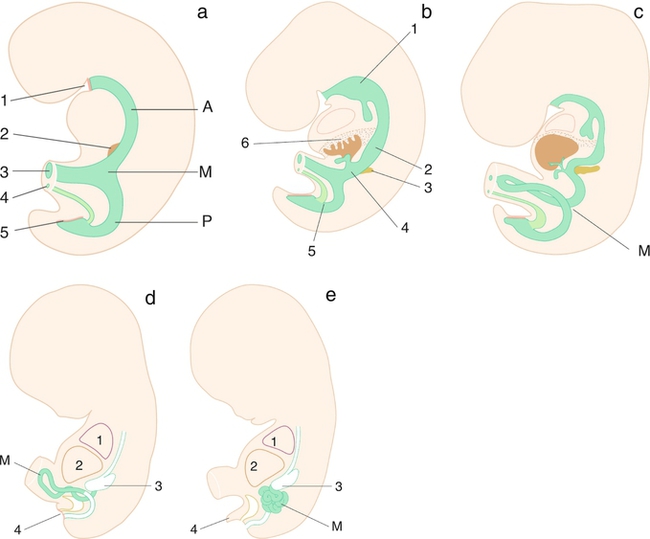
a. Embryon de 3 mm à 5 SA et 4 jours (25 jours de grossesse). Le tube digestif est fermé par la membrane pharyngienne (1) et la membrane cloacale (5). Il se divise en trois parties : intestin antérieur (A), intestin moyen (M) ou anse vitelline, intestin postérieur (P) qui se termine dans le cloaque. L’ébauche hépatique (2) apparaît à ce stade au niveau de l’intestin antérieur. Le cordon contient le canal vitellin (3) et l’allantoïde (4).
b. Embryon de 5 mm à 6 SA et 2 jours (30 jours de grossesse). L’intestin antérieur (1) s’allonge et l’arbre trachéobronchique commence à pousser sur sa face antérieure. L’estomac (2) et le duodénum (4) ne sont pas encore différenciés mais l’ébauche hépatobiliaire (6) connaît un développement très rapide alors que les bourgeons pancréatiques (3) apparaissent avec un léger retard. Au niveau du cloaque, entre l’allantoïde et l’intestin postérieur, l’éperon périnéal (5) se développe en direction de la membrane cloacale.
c. Embryon de 10 mm à 7 SA (35 jours de grossesse). L’intestin moyen (M), compris entre le troisième duodénum et le dernier tiers du côlon transverse, connaît un « extraordinaire » processus d’allongement ce qui l’oblige à se développer en dehors de la cavité abdominale, dans le cœlome du cordon : c’est la hernie ombilicale physiologique.
d. Embryon de 25 mm à 9 SA (49 jours de grossesse). Poumon (1), foie (2) et estomac (3) sont en place. L’anse intestinale primitive (M) poursuit sont développement dans le cordon. L’éperon périnéal (futur périnée) a atteint la membrane cloacale (4) qui se trouve divisée en deux autres membranes transitoires, la membrane anale en arrière et la membrane urogénitale en avant.
e. Embryon de 45 mm à 11 SA (63 jours de grossesse). L’intestin (M) a entièrement réintégré l’abdomen. En arrière du tubercule génital (4), la membrane anale et la membrane urogénitale ont régressé. Source : Tuchmann-Duplessis H, Haegel P. Embryologie. Fascicule 2 : organogenèse. Paris : Masson ; 1979.
 l’une devient intra-embryonnaire et sera à l’origine du tube digestif ;
l’une devient intra-embryonnaire et sera à l’origine du tube digestif ;
 l’autre reste extra-embryonnaire et forme la vésicule ombilicale ou vitelline.
l’autre reste extra-embryonnaire et forme la vésicule ombilicale ou vitelline.
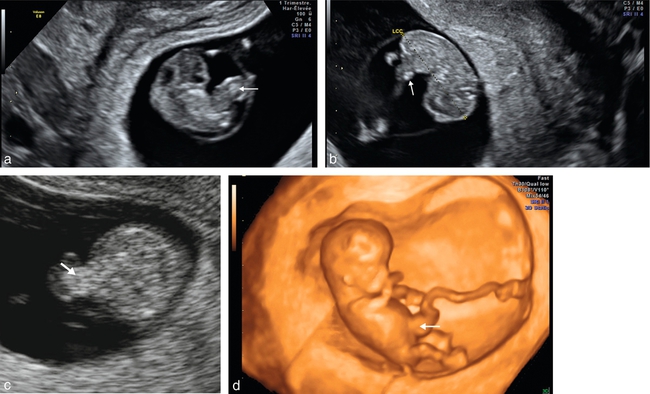
a. Coupe sagittale de l’embryon (8 SA). Le cordon est large et contient des échos denses correspondant à l’anse intestinale primitive (→).
b. Coupe sagittale de l’embryon (9 SA). On note au niveau de l’émergence du cordon ombilical la présence d’échos denses correspondant à l’anse intestinale primitive élargissant la racine du cordon (→).
c. Coupe transversale de l’abdomen passant par l’ombilic de l’embryon (11 SA). On individualise dans le pied du cordon des échos denses (→), identiques à ceux retrouvés dans l’abdomen.
d. Reconstruction 3D surface de l’embryon (12 SA). On retrouve un élargissement du cordon ombilical au niveau du nombril (→). Cet aspect est probablement encore physiologique mais mérite d’être vérifié 3 semaines plus tard pour éliminer une petite omphalocèle ou une hernie ombilicale.
 l’intestin antérieur, de la bouche au duodénum avec successivement :
l’intestin antérieur, de la bouche au duodénum avec successivement :
 l’appareil branchial qui est une succession de bourrelets de mésoblaste (les arcs branchiaux) entre lesquels se forment des poches entoblastiques qui donneront l’oreille moyenne, les amygdales, le thymus, la thyroïde,
l’appareil branchial qui est une succession de bourrelets de mésoblaste (les arcs branchiaux) entre lesquels se forment des poches entoblastiques qui donneront l’oreille moyenne, les amygdales, le thymus, la thyroïde,
 enfin, l’arbre trachéobronchique qui naît d’une évagination entoblastique antérieure au niveau de l’intestin initial (pharyngien) formant d’abord une gouttière œsophagotrachéale qui se clive secondairement de l’œsophage (voir fig. 11.1) ;
enfin, l’arbre trachéobronchique qui naît d’une évagination entoblastique antérieure au niveau de l’intestin initial (pharyngien) formant d’abord une gouttière œsophagotrachéale qui se clive secondairement de l’œsophage (voir fig. 11.1) ;
Défauts de fermeture de la paroi abdominale
 si ce sont les plis latéraux, on aura une célosomie moyenne représentée typiquement par l’omphalocèle ;
si ce sont les plis latéraux, on aura une célosomie moyenne représentée typiquement par l’omphalocèle ;
 diverses combinaisons sont possibles entre les trois formes, jusqu’à la célosomie totale.
diverses combinaisons sont possibles entre les trois formes, jusqu’à la célosomie totale.
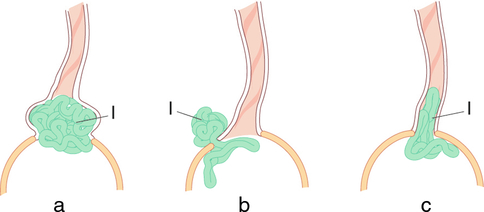
a. Omphalocèle : le cordon s’insère sur l’omphalocèle.
b. Laparoschisis : par la brèche de la paroi abdominale (à droite de l’insertion du cordon), les viscères font hernie et flottent librement dans le liquide amniotique.
c. Hernie ombilicale : le tube digestif glisse dans le cordon sans élargissement important de l’orifice ombilical.
Omphalocèle
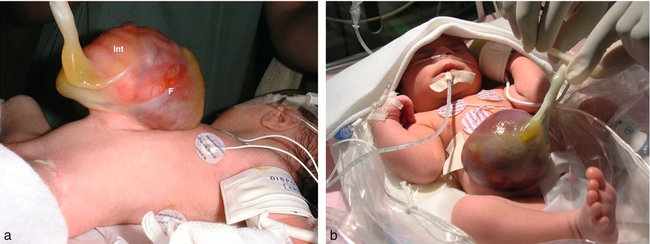
a. Omphalocèle contenant de l’intestin (Int) et une partie du foie (F).
b. Hépatocèle. Le contenu de l’omphalocèle est ici purement hépatique. Source : Dr R. Sfeir.
Étude échographique
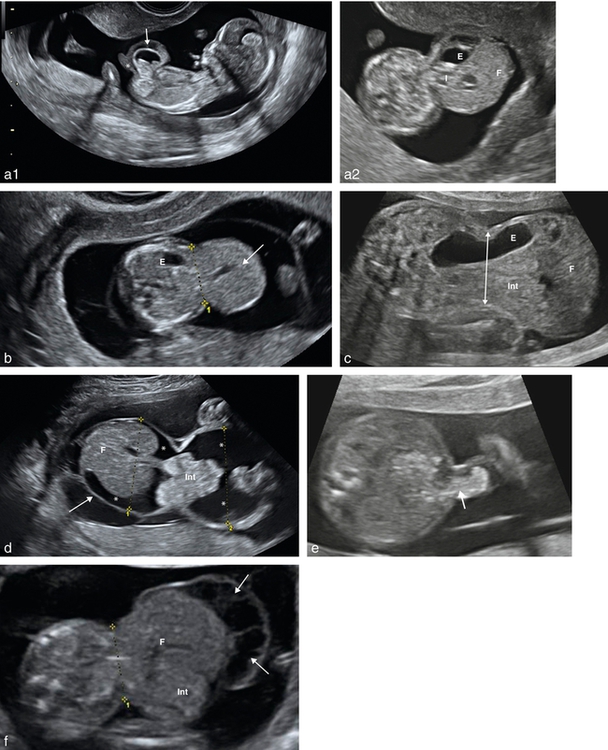
a1. 13 SA. Coupe longitudinale de l’embryon mettant en évidence une omphalocèle sus-ombilicale à contenu mixte (→). Cordon (∗). a2 La coupe transversale de l’abdomen précise ce contenu : estomac (E), intestin (I), foie (F).
b. Hépatocèle (13 SA). Coupe transversale de l’abdomen. Le contenu de l’omphalocèle est purement hépatique (→). Le collet (1) est large. L’estomac (E) est intra-abdominal.
c. 26 SA. Coupe transversale de l’abdomen. L’omphalocèle est volumineuse avec un large collet (↔). Elle comporte le foie (F), un peu d’intestin (Int) et une partie de l’estomac (E).
d. 25 SA. Coupe transversale de l’abdomen. Omphalocèle (→) dont le diamètre transverse est identique à celui de l’abdomen. Le collet est large laissant passer un peu d’intestin (Int). Le foie (F) est en totalité dans l’omphalocèle. Il existe une ascite majeure (∗).
e. 18 SA. Coupe transversale de l’abdomen passant par l’ombilic. L’intestin (→) s’insinue dans le pied du cordon au travers d’un collet de petite taille. L’étude chromosomique a révélé une trisomie 13.
f. 25 SA. Coupe transversale de l’abdomen. Cette volumineuse omphalocèle contient l’ensemble du foie (F) et de l’intestin (Int). On individualise un aspect trabéculé de la gelée de Wharton (→) à ne pas confondre avec de l’ascite.
 un élargissement de l’orifice ombilical avec, sur une coupe transversale ou sagittale de l’abdomen, la présence d’une masse bien limitée, médiane, appendue à la paroi abdominale antérieure et cernée d’une fine membrane, mobile avec les mouvements du fœtus ; la membrane est parfois épaissie, trabéculée par un épaississement localisé de la gelée de Wharton ;
un élargissement de l’orifice ombilical avec, sur une coupe transversale ou sagittale de l’abdomen, la présence d’une masse bien limitée, médiane, appendue à la paroi abdominale antérieure et cernée d’une fine membrane, mobile avec les mouvements du fœtus ; la membrane est parfois épaissie, trabéculée par un épaississement localisé de la gelée de Wharton ;
 cette masse peut contenir uniquement de l’intestin grêle échogène ou aussi une partie du foie un peu moins échogène mais homogène, parfois l’estomac et/ou la vésicule biliaire. Il est fréquent que l’omphalocèle ne comporte que du foie, parfois en totalité et l’on parlera alors d’hépatocèle (40 à 75 % des cas). La présence d’ascite est également possible sans que le pronostic de cette malformation en soit pour autant aggravé ;
cette masse peut contenir uniquement de l’intestin grêle échogène ou aussi une partie du foie un peu moins échogène mais homogène, parfois l’estomac et/ou la vésicule biliaire. Il est fréquent que l’omphalocèle ne comporte que du foie, parfois en totalité et l’on parlera alors d’hépatocèle (40 à 75 % des cas). La présence d’ascite est également possible sans que le pronostic de cette malformation en soit pour autant aggravé ;
 en Doppler couleur, la veine ombilicale contourne les viscères herniés quand il s’agit uniquement d’intestin et reste médiane à travers l’ouverture pariétale quand le foie est hernié du fait de son trajet intra-hépatique. Selon la position du cordon par rapport à l’omphalocèle, on parle ainsi d’omphalocèle centrale (cordon médian), sus-ombilicale (cordon inséré sur la partie inférieure), sous-ombilicale (cordon inséré sur la partie supérieure).
en Doppler couleur, la veine ombilicale contourne les viscères herniés quand il s’agit uniquement d’intestin et reste médiane à travers l’ouverture pariétale quand le foie est hernié du fait de son trajet intra-hépatique. Selon la position du cordon par rapport à l’omphalocèle, on parle ainsi d’omphalocèle centrale (cordon médian), sus-ombilicale (cordon inséré sur la partie inférieure), sous-ombilicale (cordon inséré sur la partie supérieure).
Diagnostic différentiel
 Lorsque le contenu est purement intestinal (30 % des cas) et de petit volume, on peut discuter le diagnostic de hernie ombilicale où dans un ombilic large s’extériorise au minimum une anse intestinale très échogène, mais la peau et l’insertion du cordon sont normales (fig. 13.6a). Les examens complémentaires à proposer seront les mêmes que pour une omphalocèle intestinale.
Lorsque le contenu est purement intestinal (30 % des cas) et de petit volume, on peut discuter le diagnostic de hernie ombilicale où dans un ombilic large s’extériorise au minimum une anse intestinale très échogène, mais la peau et l’insertion du cordon sont normales (fig. 13.6a). Les examens complémentaires à proposer seront les mêmes que pour une omphalocèle intestinale.
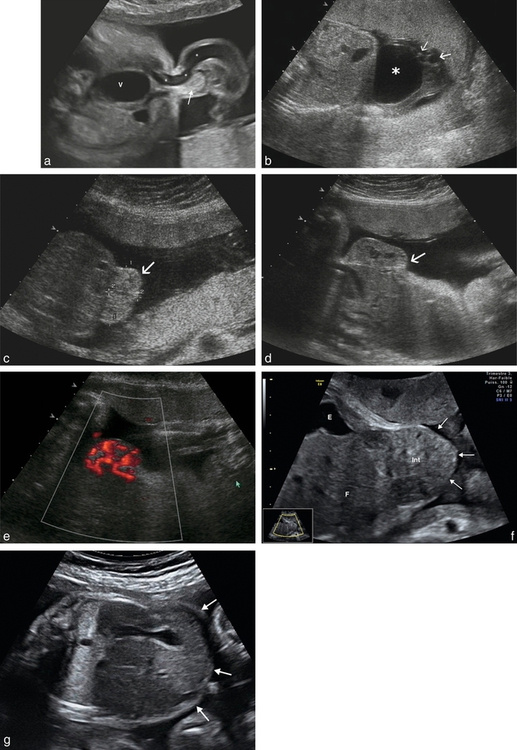
a. Hernie ombilicale (26 SA). Coupe transversale de l’abdomen au niveau de l’ombilic, légèrement inclinée vers le bas. L’ombilic est large avec à côté de la veine ombilicale (∗) une petite hernie intestinale caractérisée par une masse échogène dans le pied du cordon (→). V : vessie.
b. Kyste allantoïdien : image liquidienne (∗) accolée à la paroi abdominale antérieure, avec une paroi épaisse formée de gelée de Wharton contenant les vaisseaux ombilicaux (→).
c. Hémangiome péri-ombilical (de type RICH) à 27 SA : en coupe transversale, tumeur échogène (→) de 25 × 15 mm faisant saillie dans la région ombilicale, pouvant évoquer une célosomie.
d. Hémangiome péri-ombilical (→) en coupe sagittale, même fœtus qu’en c : aspect lacunaire de la tumeur (35 × 15 mm) et la paroi abdominale semble intacte.
e. Hémangiome péri-ombilical : sauvé par le Doppler couleur ! L’hypervascularisation permet d’évoquer une tumeur vasculaire et donc l’hémangiome congénital.
f. Éventration (37 SA). Coupe transversale de l’abdomen fœtal. Celui-ci est modelé par l’utérus. On note une protrusion antérieure de la paroi abdominale fine et régulière (→). Il s’agit d’une éventration secondaire à un gros kyste abdominal persistant au début du 2e trimestre (sans diagnostic étiologique précis) et affaissé par ponction vers 18 SA. Un défaut de développement musculo-aponévrotique abdominal n’a pas pu être évité. E : estomac ; F : foie ; Int : intestin.
g. Faux aspect d’omphalocèle (28 SA). Cette coupe transversale de l’abdomen est un peu trop oblique et le modelage de l’abdomen par l’utérus et les membres du fœtus crée un faux aspect d’omphalocèle. On note que la paroi de l’abdomen (→) est d’épaisseur habituelle, ce qui est en faveur de sa structure normale.
 Une lésion kystique ombilicale peut évoquer un kyste allantoïdien (fig. 13.6b), un kyste du canal omphalomésentérique, un épaississement localisé de la gelée de Wharton, un hémangiome du cordon.
Une lésion kystique ombilicale peut évoquer un kyste allantoïdien (fig. 13.6b), un kyste du canal omphalomésentérique, un épaississement localisé de la gelée de Wharton, un hémangiome du cordon.
 Les tumeurs de la paroi abdominale antérieure, rares, peuvent simuler une célosomie moyenne, mais l’analyse va montrer l’intégrité de la paroi musculaire. Il s’agit surtout d’angiomes congénitaux (plutôt de type RICH pour rapid involuting congenital hemangioma et de bon pronostic), échogènes, que le Doppler couleur aide à identifier (fig. 13.6c, d et e).
Les tumeurs de la paroi abdominale antérieure, rares, peuvent simuler une célosomie moyenne, mais l’analyse va montrer l’intégrité de la paroi musculaire. Il s’agit surtout d’angiomes congénitaux (plutôt de type RICH pour rapid involuting congenital hemangioma et de bon pronostic), échogènes, que le Doppler couleur aide à identifier (fig. 13.6c, d et e).
 Une exceptionnelle éventration abdominale peut être la conséquence d’une distension abdominale précoce parfois résolutive (mégavessie, kyste abdominal) suffisante pour altérer la formation pariétale (équivalent de syndrome de prune belly) (fig. 13.6f).
Une exceptionnelle éventration abdominale peut être la conséquence d’une distension abdominale précoce parfois résolutive (mégavessie, kyste abdominal) suffisante pour altérer la formation pariétale (équivalent de syndrome de prune belly) (fig. 13.6f).
 Un modelage de la paroi abdominale sur le placenta, sur le myomètre ou en cas d’oligoamnios peut créer un faux aspect d’omphalocèle (fig. 13.6g).
Un modelage de la paroi abdominale sur le placenta, sur le myomètre ou en cas d’oligoamnios peut créer un faux aspect d’omphalocèle (fig. 13.6g).
 Une omphalocèle rompue donne un aspect de laparoschisis (voir plus bas).
Une omphalocèle rompue donne un aspect de laparoschisis (voir plus bas).
Associations malformatives et anomalies chromosomiques
Malformations associées
 malformations cardiaques (20 à 50 %) surtout dans les omphalocèles volumineuses et supérieures, rarement dans les célosomies inférieures : tétralogie de Fallot, communication interventriculaire, communication interauriculaire, canal atrioventriculaire, cardiopathies complexes ;
malformations cardiaques (20 à 50 %) surtout dans les omphalocèles volumineuses et supérieures, rarement dans les célosomies inférieures : tétralogie de Fallot, communication interventriculaire, communication interauriculaire, canal atrioventriculaire, cardiopathies complexes ;
 malformations génito-urinaires (17 %), essentiellement en cas d’association avec une célosomie inférieure (exstrophies vésicale ou cloacale, voir plus loin et fig. 14.39 et 14.40) : dysplasies rénales, méga-uretère, reins polykystiques ;
malformations génito-urinaires (17 %), essentiellement en cas d’association avec une célosomie inférieure (exstrophies vésicale ou cloacale, voir plus loin et fig. 14.39 et 14.40) : dysplasies rénales, méga-uretère, reins polykystiques ;
 malformations du système nerveux central (7 %) et de la face (16 %) : anencéphalie, microcéphalie, fentes labiopalatines, myéloméningocèle ;
malformations du système nerveux central (7 %) et de la face (16 %) : anencéphalie, microcéphalie, fentes labiopalatines, myéloméningocèle ;
 malformations des membres et des extrémités (6 %) : polydactylie, syndactylie, pied bot, amélie ;
malformations des membres et des extrémités (6 %) : polydactylie, syndactylie, pied bot, amélie ;
 malformations intestinales paradoxalement beaucoup plus rares (1 %) : atrésies intestinales, diverticule de Meckel.
malformations intestinales paradoxalement beaucoup plus rares (1 %) : atrésies intestinales, diverticule de Meckel.
 le syndrome de Beckwith-Wiedemann, dont la fréquence est estimée à 1/14 000 naissances, se caractérise par une croissance excessive du fœtus et associe de façon variable : omphalocèle (75 % des cas), macroglossie, macrosomie avec viscéromégalie, hépatosplénomégalie (32 %) ou néphromégalie (23 %), cardiopathies (15 %). De nombreuses autres anomalies sont plus rarement retrouvées : microcéphalie, dysmorphie faciale, exophtalmie, occiput proéminent, hémi-hypertrophie totale ou partielle, hernie diaphragmatique, malformation génito-urinaire, tumeur hépatique, rénale (tumeur de Wilms) ou surrénalienne, hémangiome ou placenta kystique. La complication néonatale essentielle est l’hypoglycémie secondaire à un hyperinsulinisme. La mortalité est de 20 %, secondaire à cette hypoglycémie mais aussi aux complications des malformations associées. Le gène est identifié : ce syndrome est secondaire à une anomalie sur le locus p15.5 du chromosome 11. La majorité des cas sont sporadiques (85 %), les autres cas sont des formes familiales ;
le syndrome de Beckwith-Wiedemann, dont la fréquence est estimée à 1/14 000 naissances, se caractérise par une croissance excessive du fœtus et associe de façon variable : omphalocèle (75 % des cas), macroglossie, macrosomie avec viscéromégalie, hépatosplénomégalie (32 %) ou néphromégalie (23 %), cardiopathies (15 %). De nombreuses autres anomalies sont plus rarement retrouvées : microcéphalie, dysmorphie faciale, exophtalmie, occiput proéminent, hémi-hypertrophie totale ou partielle, hernie diaphragmatique, malformation génito-urinaire, tumeur hépatique, rénale (tumeur de Wilms) ou surrénalienne, hémangiome ou placenta kystique. La complication néonatale essentielle est l’hypoglycémie secondaire à un hyperinsulinisme. La mortalité est de 20 %, secondaire à cette hypoglycémie mais aussi aux complications des malformations associées. Le gène est identifié : ce syndrome est secondaire à une anomalie sur le locus p15.5 du chromosome 11. La majorité des cas sont sporadiques (85 %), les autres cas sont des formes familiales ;
 la pentalogie de Cantrell (fig. 13.7) entre dans le cadre des célosomies supérieures :
la pentalogie de Cantrell (fig. 13.7) entre dans le cadre des célosomies supérieures :
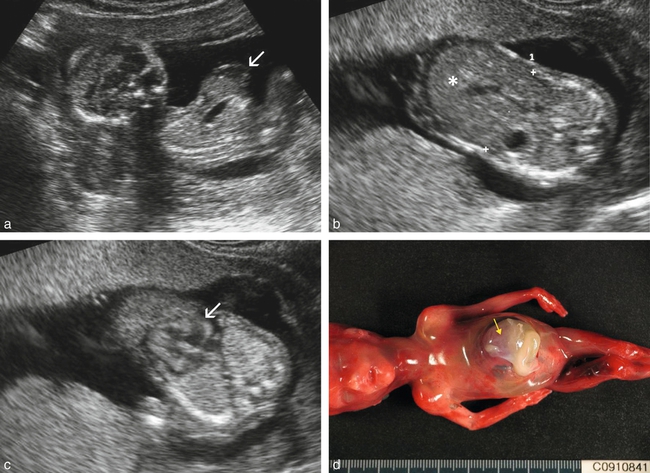
a. Aspect de célosomie avec extension thoracique (→).
b. Coupe transversale de l’abdomen : volumineuse omphalocèle (hépatocèle ∗).
c. Coupe transversale dans le bas du thorax : ectopie cardiaque (→) partielle (le cœur est engagé dans l’omphalocèle).
d. Aspect du fœtus. Le cœur (→) est visible à la partie haute de l’omphalocèle. Source fig. 13.7d : Dr L. Devisme.
 ce syndrome polymalformatif exceptionnel, dans la plupart des cas sporadique, dépisté dès le 1er trimestre de grossesse, associe dans sa forme complète :
ce syndrome polymalformatif exceptionnel, dans la plupart des cas sporadique, dépisté dès le 1er trimestre de grossesse, associe dans sa forme complète :
 ce syndrome est à différencier de :
ce syndrome est à différencier de :
 le syndrome OEIS, exceptionnel (1/200 000 naissances), associe une omphalocèle, une exstrophie vésicale, une imperforation anale et un spina bifida (large myéloméningocèle lombo-sacrée) visualisé directement ou suspecté sur les signes intracrâniens (malformation de Chiari). Il peut s’y associer une scoliose, des pieds bots et un thorax étroit (voir chap. 10 et fig. 14.40) ;
le syndrome OEIS, exceptionnel (1/200 000 naissances), associe une omphalocèle, une exstrophie vésicale, une imperforation anale et un spina bifida (large myéloméningocèle lombo-sacrée) visualisé directement ou suspecté sur les signes intracrâniens (malformation de Chiari). Il peut s’y associer une scoliose, des pieds bots et un thorax étroit (voir chap. 10 et fig. 14.40) ;
 le syndrome du cordon court (fig. 13.8). D’une fréquence estimée à 1/30 000 grossesses, il est probablement plus fréquent car il est responsable d’avortements spontanés avant le diagnostic échographique. Le cordon ombilical paraît court voire absent (recherché en Doppler couleur). Les viscères abdominaux sont extériorisés dans l’espace extracœlomique (cœlome externe) et sont accolés plus ou moins étroitement à la surface placentaire. Ce syndrome associe de larges defects crâniens, une malformation de la paroi thoracique et/ou abdominale, des amputations des membres, une cyphoscoliose, des malformations viscérales et un oligoamnios. Il n’est généralement pas retrouvé d’anomalie chromosomique. On en rapproche le limb body wall complex (ou syndrome) : séquence cordon court (abdominoschisis, scoliose) ± exencéphalie, fente labiale, ectromélie, imperforation anale ± anomalies génitales ± myéloméningocèle (voir chap. 10). On peut aussi en rapprocher la grossesse extra-membraneuse (voir chap. 5) et le syndrome des brides amniotiques qui associe, dans sa forme complexe, asymétrie faciale avec exencéphalie, amputations des doigts et des orteils, abdominochisis.
le syndrome du cordon court (fig. 13.8). D’une fréquence estimée à 1/30 000 grossesses, il est probablement plus fréquent car il est responsable d’avortements spontanés avant le diagnostic échographique. Le cordon ombilical paraît court voire absent (recherché en Doppler couleur). Les viscères abdominaux sont extériorisés dans l’espace extracœlomique (cœlome externe) et sont accolés plus ou moins étroitement à la surface placentaire. Ce syndrome associe de larges defects crâniens, une malformation de la paroi thoracique et/ou abdominale, des amputations des membres, une cyphoscoliose, des malformations viscérales et un oligoamnios. Il n’est généralement pas retrouvé d’anomalie chromosomique. On en rapproche le limb body wall complex (ou syndrome) : séquence cordon court (abdominoschisis, scoliose) ± exencéphalie, fente labiale, ectromélie, imperforation anale ± anomalies génitales ± myéloméningocèle (voir chap. 10). On peut aussi en rapprocher la grossesse extra-membraneuse (voir chap. 5) et le syndrome des brides amniotiques qui associe, dans sa forme complexe, asymétrie faciale avec exencéphalie, amputations des doigts et des orteils, abdominochisis.
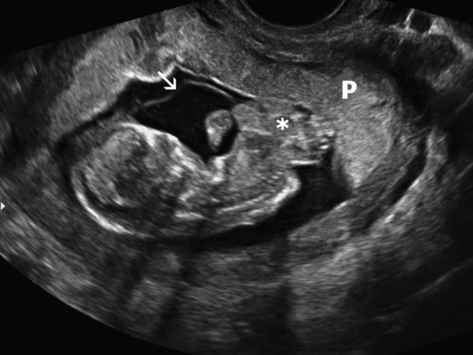
13 SA. Éviscération précoce (abdominoschizis) : les viscères abdominaux sont extériorisés (∗) dans le cœlome externe et sont accolés plus ou moins étroitement à la surface placentaire (P). La membrane amniotique est encore bien visible (→) et souligne la situation extra-amniotique des viscères : il s’agit ici d’une anomalie très précoce de la morphogenèse, au moment de la délimitation embryonnaire.
Anomalies chromosomiques
 si l’omphalocèle est petite, ne comportant que des anses intestinales (87 % si le foie est intra-abdominal contre 9 % quand le foie est extériorisé) ;
si l’omphalocèle est petite, ne comportant que des anses intestinales (87 % si le foie est intra-abdominal contre 9 % quand le foie est extériorisé) ;
 s’il existe une association avec un kyste du cordon (pseudo-kyste ou kyste allantoïdien) ;
s’il existe une association avec un kyste du cordon (pseudo-kyste ou kyste allantoïdien) ;
 ou s’il existe la notion d’une hyperclarté nucale au 1er trimestre (voir chap. 16).
ou s’il existe la notion d’une hyperclarté nucale au 1er trimestre (voir chap. 16).
Laparoschisis
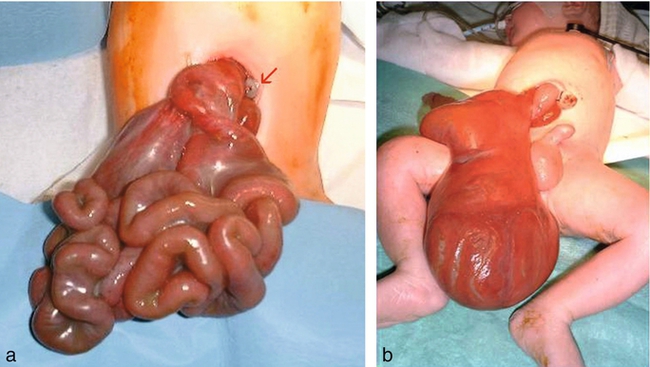
a. Orifice bien limité à droite de l’origine (→) du cordon. Pas de sac herniaire. On note la dilatation de certaines anses, l’inflammation à proximité de l’orifice et l’agglutination avec accolement témoignant d’une périviscérite (péritonite « chimique » in utero).
b. Ce nouveau-né est porteur d’un laparoschisis compliqué de périviscérite. Les anses intestinales sont agglutinées et entourées d’une enveloppe épaisse. Source : Dr R. Sfeir.
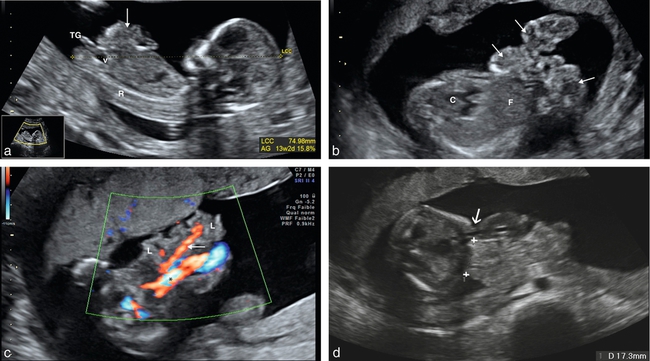
a. Coupe sagittale de l’embryon. 13 SA. La paroi abdominale est anormale permettant l’extériorisation des anses intestinales (→). On note que la vessie (v) est en place chez cet embryon de sexe féminin : le tubercule génital (TG) est parallèle au rachis (R).
b. 19 SA. Coupe oblique thoraco-abdominale. Les anses intestinales flottent dans le liquide amniotique (→). Elles se détachent nettement de la paroi abdominale. La lumière intestinale ne se distingue pas encore. C : cœur ; F : foie.
c. 16 SA. Coupe transversale de l’abdomen passant par l’ombilic avec Doppler couleur. On distingue à côté du cordon ombilical (∗), individualisé par les artères ombilicales, l’artère mésentérique supérieure (→) qui vient vasculariser le laparoschisis (L).
d. 23 SA. L’intestin sort à droite du cordon (→). Mesure du collet dont le diamètre dépasse les 15 mm, plutôt de bon pronostic.
 la présence de plusieurs anses intestinales, herniées à travers un orifice para-ombilical droit (dont on mesure le diamètre), et flottant librement dans le liquide amniotique, sans membrane recouvrant la malformation ;
la présence de plusieurs anses intestinales, herniées à travers un orifice para-ombilical droit (dont on mesure le diamètre), et flottant librement dans le liquide amniotique, sans membrane recouvrant la malformation ;
 plus rarement, d’autres organes peuvent s’extérioriser comme l’estomac, le foie, la vésicule biliaire, la vessie. Toutefois, la présence d’une éviscération de ces organes doit faire craindre une pathologie plus complexe (syndrome du cordon court, maladie des brides amniotiques, exstrophie cloacale) et faire rechercher d’autres malformations associées.
plus rarement, d’autres organes peuvent s’extérioriser comme l’estomac, le foie, la vésicule biliaire, la vessie. Toutefois, la présence d’une éviscération de ces organes doit faire craindre une pathologie plus complexe (syndrome du cordon court, maladie des brides amniotiques, exstrophie cloacale) et faire rechercher d’autres malformations associées.
 le diamètre des anses herniées : la lumière intestinale du grêle est considérée comme dilatée au-delà de 6 mm, ce qui est souvent prédictif de complications post-natales. Au-delà de 17 mm, la morbidité est élevée avec notamment un risque d’atrésie du grêle. Mais l’état de l’intestin intra-abdominal est également important : une dilatation « asymétrique » des anses grêles intra-abdominales sans dilatation des anses éviscérées est également de mauvais pronostic (fig. 13.11a et b) ;
le diamètre des anses herniées : la lumière intestinale du grêle est considérée comme dilatée au-delà de 6 mm, ce qui est souvent prédictif de complications post-natales. Au-delà de 17 mm, la morbidité est élevée avec notamment un risque d’atrésie du grêle. Mais l’état de l’intestin intra-abdominal est également important : une dilatation « asymétrique » des anses grêles intra-abdominales sans dilatation des anses éviscérées est également de mauvais pronostic (fig. 13.11a et b) ;
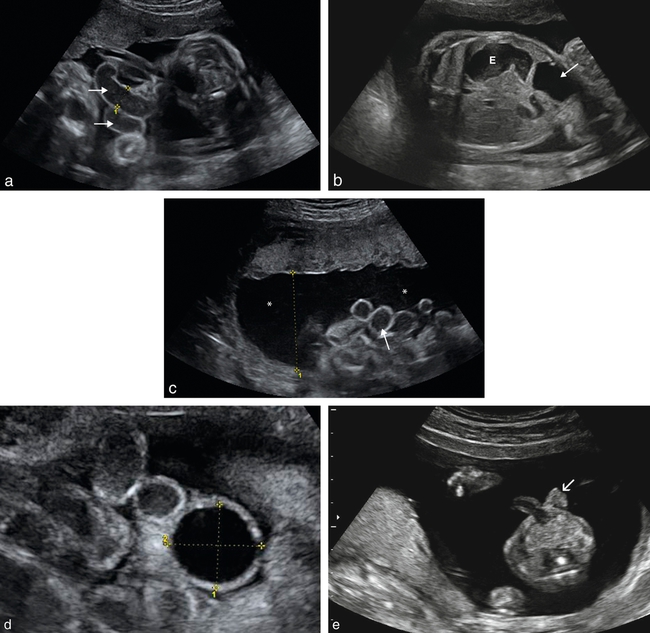
a. Les anses digestives extériorisées s’individualisent aisément (→). La lumière intestinale (1) est mesurée à 13 mm, ce qui est une mesure large. La paroi digestive, en revanche, est d’épaisseur normale. Le contenu est d’échogénicité normale.
b. 23 SA. Coupe transversale de l’abdomen. L’estomac (E) est bien visible et on retrouve sur ce plan de coupe la dilatation d’au moins une anse digestive intra-abdominale (→).
c. Le liquide amniotique (∗) est abondant en raison du syndrome obstructif intra-abdominal haut qui a compliqué le laparoschisis (→).
d. 32 SA. On individualise une anse intestinale très dilatée (20 mm) avec une paroi épaissie (2 à 3 mm).
e. 18 SA. Coupe transversale de l’abdomen. On retrouve à la droite de l’ombilic un petit amas d’anses intestinales sans lumière intestinale visible (→). Le collet très petit ne peut être vu. La striction sur l’intestin est tellement importante qu’elle entraînera une nécrose totale puis la disparition de l’intestin extra-abdominal (vanishing gut).
 l’apparition d’un hydramnios (fig. 13.11c) ;
l’apparition d’un hydramnios (fig. 13.11c) ;
 l’épaississement et l’échogénicité de la paroi intestinale : au-delà de 3 mm, il s’accompagne d’une plus grande morbidité post-natale (fig. 13.11d) ;
l’épaississement et l’échogénicité de la paroi intestinale : au-delà de 3 mm, il s’accompagne d’une plus grande morbidité post-natale (fig. 13.11d) ;
 la taille de l’orifice pariétal : une ouverture pariétale étroite (inférieure à 10 mm) augmente le risque d’ischémie intestinale par strangulation des anses et du mésentère (nécrose totale intestinale extra-abdominale ou vanishing gut) (fig. 13.11e) ;
la taille de l’orifice pariétal : une ouverture pariétale étroite (inférieure à 10 mm) augmente le risque d’ischémie intestinale par strangulation des anses et du mésentère (nécrose totale intestinale extra-abdominale ou vanishing gut) (fig. 13.11e) ;
 la vascularisation des anses herniées : il est possible de l’apprécier en recherchant un signal Doppler au niveau de la paroi digestive, notamment par Doppler énergie, et en mesurant l’index de résistance artérielle ou de pulsatilité de l’artère mésentérique supérieure dans sa portion herniée et dans sa portion intra-abdominale au niveau de son origine et de l’ouverture pariétale.
la vascularisation des anses herniées : il est possible de l’apprécier en recherchant un signal Doppler au niveau de la paroi digestive, notamment par Doppler énergie, et en mesurant l’index de résistance artérielle ou de pulsatilité de l’artère mésentérique supérieure dans sa portion herniée et dans sa portion intra-abdominale au niveau de son origine et de l’ouverture pariétale.
Exstrophie vésicale
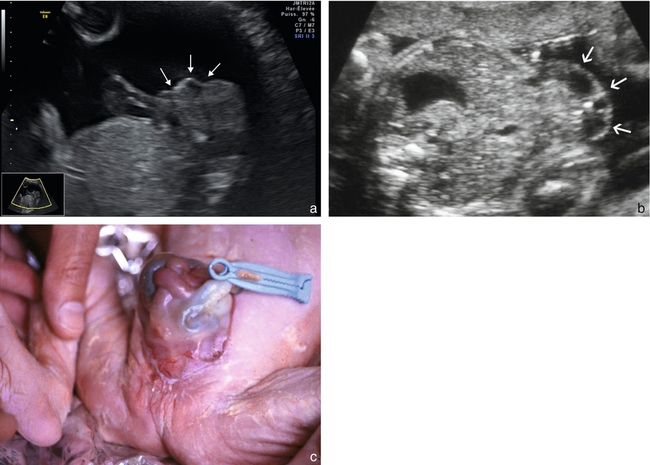
a. Exstrophie vésicale (23 SA). Coupe sagittale abdominopelvienne du fœtus. Paroi abdominale irrégulière et bourgeonnante (→) en dessous de l’implantation du cordon qui paraît basse. La vessie n’est pas vue. La quantité de liquide amniotique est habituelle.
b. Célosomie inférieure (27 SA). Coupe abdominale un peu oblique vers le bas. On distingue une paroi antérieure irrégulière et bourgeonnante en raison d’une omphalocèle inférieure comportant quelques anses grêles (→). En dessous de cette omphalocèle existe une exstrophie vésicale (non visible sur ce cliché).
c. Nouveau-né de l’écho en b : célosomie inférieure avec petite ouverture vésicale. Il existait aussi une imperforation anale.![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


13: Pathologie de la paroi et du contenu abdominal













